


INTRODUCTION
Primary pulmonary hypertension (PPH) is an uncommon disease that affects predominantly young and productive people1–3). Although important conceptual advances and information about the pathogenesis, histopathology, characterization and management of PPH have been generated in the past decade, many questions remain to be answered. Part of the reason for the lack of complete knowledge in this regard is a rare entity and a short mean survival of 2 to 4 years2–5). Patients with a survival period > 5 to 10 years have also been well documented5–9) and even spontaneous regression of the disease, although uncommon, has been described10–11). We analyzed determinants associated with the survival and prognosis of patients with PPH.
MATERIALS AND METHODS
1. Materials
The study series included 13 patients in whom PPH was diagnosed at our institution between 1988 and 1996 and who were followed-up through July 1999. As in other studies, the PPH was diagnosed by a thorough workup, including clinical history, physical examination, laboratory tests, chest radiography, ECG, pulmonary function tests, echocardiography, radionuclide perfusion lung scan and cardiac catheterization. Criteria used to establish the diagnosis of PPH include a mean pulmonary arterial pressure of more than 25 mmHg at rest or 30 mmHg during exercise, a normal pulmonary capillary wedge pressure (PCWP) and absence of other disease known to cause or to be associated with secondary pulmonary hypertension12). Particular care was taken to exclude patients with evidence of congenital heart disease or acquired vascular or myocardial disease, obstructive or restrictive lung disease, or both, parasitic disease involving the lung, pulmonary thromboembolic and clearly defined collagen vascular disease and the antiphospholipid syndrome.
2. Hemodynamic measurements
Cardiac output and cardiac index were measured by the thermodilution method and pulmonary artery pressure, PCWP, right atrial pressure, right ventricular pressure and oxygen saturation were obtained by a Swan-Ganz catheter. The radial artery was also cannulated with a needle for arterial oxygen blood sampling. We did not evaluate the hemodynamic data after taking a vasodilator drug during cardiac catheterization.
3. Treatment
Some patients in this study had been taking a vasodilator and diuretics. All patients were not treated with coumadin.
4. Statistical analysis
For the survival analysis, we used the initial clinical and hemodynamic characteristics by cardiac catheterization as an index for determining survival. All values are expressed as mean ± SD. The Kaplan-Meier method was used to estimate overall survival distribution and Cox proportional hazards model was used to examine the relation between survival and variables. A p value <0.05 was significant in all analysis.
RESULTS
1. Clinical characteristics
The mean age of the patients with PPH entered into the study was 36.1 ± 9.3 years and women were predominant (female/male ratio 1.75:1). None of the patients had histories of appetite suppressant drug use and 3 were cigarette smokers. Of the women of reproductive age, one had taken oral contraceptive and none had a family history of familial pulmonary hypertension. The frequency of symptoms at diagnosis was dyspnea 100%, syncope 2 (16.7%), chest pain 2 (16.7%), effort related palpitation 1 (8.3%), hoarseness 4 (30.7%) and leg edema 3 (23.1%). The functional status of the patients at diagnosis according to the New York Heart Association (NYHA) classification was as follows; 8.3% of the patients were in class I, 41.7% in class II, 41.7% in class III and 16.7% in class IV (Table 1). Mean time from onset of the first symptom to diagnosis of PPH was 4.2 years (range 0 to 9.6).
2. Laboratory findings
The chest radiograph showed the typical changes associated with pulmonary hypertension, namely prominence of the main pulmonary artery in all patients and enlarged hilar vessels. The mean cardiothoracic ratio was 0.6. The ECG showed a sinus rhythm and evidence of right ventricular hypertrophy in all cases. The echocardiogram confirmed right ventricular hypertrophy and showed variable degrees of right ventricular enlargement. A lung perfusion scan was performed in 8 patients and was considered to be normal in 4 (50%) and showed diffuse bilateral patchy pattern in 4 (50%). The mean hemoglobin and hematocrit levels were 13.9 ± 5 g/dL and 41.7 ± 4.5%, respectively. The total platelet count was normal. The antinuclear antibody test was negative in all cases.
4. Factors associated with survival and prognosis
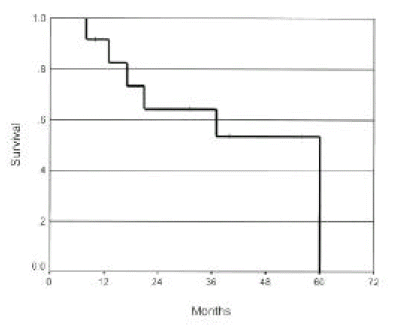
As of July 1999, 7 of the 13 PPH patients have died, and the mean survival time of this group was 3.4 ± 0.6 years. Among the 7 patients one survived <1 year, 3 < 2 years and 3 > 3 years (Figure 1). Mortality was not associated with age, sex, functional status (NYHA) and pulmonary function test. Only cardiac index was associated with the prognosis (Table 3).
DISCUSSION
PPH was first described over 100 years ago in a patient with right-heart failure whose necropsy showed no obvious reason for pulmonary arteriosclerosis13). PPH is clinically defined as a mean pulmonary arterial pressure of more than 25 mmHg at rest or 30 mmHg during exercise without evidence of secondary pulmonary hypertension14–15). The estimated annual incidence of PPH in European and US studies is 1–2 cases per one million people per year in the general population, and necropsy studies have shown a prevalence of 1300 per million16). The incidence of PPH among users of appetite suppressants may be as high as 25–50 per million per year17). The mean age at diagnosis of PPH is 36 years and the female is means predominant (1.7–3.5) than the male18). Familial PPH accounts for roughly 10% of cases. We had similar results showing the mean age was 36 years and the female was in excess (1.75) of the male. The pathogenesis of PPH envisage individual susceptibility, triggering a stimulus as the initiating factor for pulmonary vascular injury and repair, and genetic susceptibility in case of familial PPH. The stimuli that trigger PPH are drugs such as appetite suppressants, infections particularly HIV-1, and inflammatory disorders. Circulating vasoactive mediators (serotonin) may play a part in pulmonary hypertension19). The earliest symptom in most cases of PPH is the gradual onset of shortness of breath after physical exertion. Other symptoms include chest pain, syncope, fatigue, leg edema and hoarseness. We found 4 cases (30.7%) of PPH whose initial symptom was hoarseness. So, in the case of patients with hoarseness, PPH should be considered a differential diagnosis. The signs of PPH are an accentuated second heart sound in the pulmonary region, and a right ventricular S4 gallop. The aim of diagnostic testing in patients with suspected PPH is to exclude secondary causes of pulmonary hypertension. At initial screening, blood tests should include liver function tests and assays for antibodies to HIV-1, and serological studies should aim to exclude occult collagen vascular disease. Pulmonary function tests should be done to exclude significant parenchyma or airway disorders. Patients with severe PPH may have a mild restrictive pattern. Cardiac catheterization is the most important test in the assessment of pulmonary hypertension. Catheterization is necessary to fully assess right and left heart hemodynamics, the presence of shunts and vasoreactivity during acute drug trials. Acute vasodilator testing is an important component of the hemodynamic assessment, since the responses to acute challenge with vasodilators is predictive of the long-term response to oral vasodilator therapy20). We did not perform the vasodilator testing during the cardiac catheterization. Almost every type of vasodilator has been tried in the past, but there have been no prospective randomized trials of oral vasodilator therapy for PPH. Non-controlled studies have shown improved hemodynamics, exercise tolerance and survival in some patients treated with oral vasodilators20). A therapy using a high dose of calcium channel antagonists that are titrated to the maximal response of the pulmonary artery pressure and pulmonary vascular resistance has been described, and associated with a dramatic improvement in quality of life and lifestyle, regression of right ventricular hypertrophy and improved survival21). Continuous intravenous epoprostenol (prostacyclin, PGI2) has been shown to improve hemodynamics, to improve tolerance of exercise and to prolong survival in severe PPH22–24). Two trials have suggested improved survival for PPH patients treated with anticoagulants4,21). The prognosis for untreated PPH is poor. In a series of 137 cases from the UK, the median survival time was 3.4 years25). Among 200 patients enrolled on the US National Institute of Health Registry, the mean life expectancy was 2.5 years from diagnosis of PPH. This study showed 64% survival at 1 year and 48% survival at 3 years. The results were not affected by age, age at onset, sex, symptom duration, a positive test for antinuclear antibodies, family history, use of oral contraceptives, pregnancy or smoking status25). Stroke volume index, cardiac index, right atrial pressure and mean pulmonary artery pressure at catheterization are linked to survival7). Patients who respond to chronic therapy with calcium-channel blockers have a 95% chance of a 5-year life-expectancy when anticoagulant therapy is used at the same time21). Epoprostenol has increased survival in patients who are unresponsive to oral vasodilators, and is associated with a 5-year survival comparable with or better than survival after lung transplantation26). In regard to our results, the mean survival time was 3.4 years and the cardiac index is only associated with survival.
The limitation of this study includes that we have tried a common conventional vasodilator drug without a test of the vasodilator during the cardiac catheterization, so we excluded the drug for prognostic factor of survival. Not all patients have taken anticoagulation and prostacyclin. Although, there are a small number of cases and incomplete data of our study, no data has been published about survival and prognostic factor in all patients with PPH in Korea. Multicenter trial is needed to evaluate the survival and prognostic factor in all patients with PPH in Korea. In conclusion, patients with PPH have a poor survival expectancy and in this limited study with a small number of patients, mortality is largely associated with decreased cardiac index.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print





