


INTRODUCTION
Adult onset StillŌĆÖs disease (AOSD) is a chronic systemic inflammatory disorder in which high spiking fever, typical skin rash, and polyarthritis occur. The main biological features are neutrophilic leukocytosis, hyperferritinemia, and negative rheumatoid factor (RF) or antinuclear antibodies (ANA). Others may include splenomegaly, pleuritis, pericarditis, and hepatic abnormalities1). Even though functional prognosis essentially depends on articular involvement, life-threatening prognosis depends on serious complications, such as hepatic failure, disseminated intravascular coagulopathy, hemophagocytosis, infections, amyloidosis, and cardiomyopathy2ŌĆō5). In this article, we describe a patient with AOSD who had secondary renal amyloidosis. We suggest a successful combined therapy of prednisolone (PD), colchicine (COL), and cyclophosphamide (CTX) and review the literature.
CASE
A 25-year-old Korean woman was diagnosed with AOSD four years ago after experiencing a high spiking fever, maculopapular rash, and polyarthritis in her hands, elbows and knees. In laboratory findings, the leukocyte count was 19,900/ ╬╝L, the serum ferritin level was 719.3 ng/mL (10ŌĆō240), and RF and ANA were negative. During a follow-up, typical skin rash had disappeared after administration of PD, sulfasalazine or hydroxychloroquine and methotraxate (MTX), but either high fever or polyarthitis was wax and wane, and occasionally, intra-articular injections of corticosteroid were administered. In July 1998, she was admitted to our hospital because of slowly increasing proteinuria over a 7 month period without pitting edema or hypertension. She was single and had no family history of any rheumatic disease or drug history, such as gold or D-penicilliamine.
The results of laboratory data showed that the white-cell count (WBC) was 12,800 / ╬╝L, hemoglobin (Hb) was 10.6 g/dL, platelet was 610,000 / ╬╝L, ESR was 61 mm/hr, and C-reactive protein (CRP) was 9.50 mg/dL (<0.8). The serum protein and albumin had decreased to 4.5 g/dL (6.4ŌĆō8.5) and 2.1 g/dL (3.2ŌĆō5.5), respectively. Serum ferritin was within the normal range. Serum autoantibodies and rheumatoid factor were persistently negative. There was no evidence of infection or malignancy. Several urine examinations showed proteinuria without any abnormality in urinary sediments. Twenty-four hour urine protein excretion was 6.9 g/day, and urine creatine clearance was 93.3 mL/min. In radiographic findings, chest PA was normal and bony erosions were detected in both wrist joints. In the sonography, the sizes of both kidneys were 11.5cm on the right and 11cm on the left side with increased renal parenchymal echogenicity.
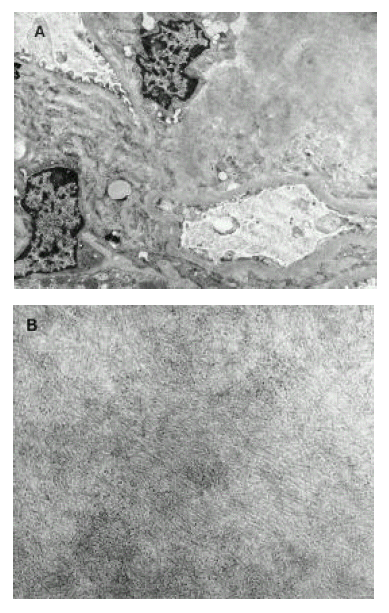
Renal biopsy showed amorphous deposits in the mesangial areas, some glomerular capillary walls and in the vascular poles (Figure 1). The amyloid deposit stained with Congored displayed apple-green birefringence under the polarizing light. Immunofluorescence study for IgG, IgM, IgA, C3, C1, C4, fibrinogen, albumin, and light chains showed segmental positive staining for IgM and C3 in the mesangium and trace (┬▒) positive staining for light chain. Electron microscopic examination confirmed amyloid deposits in the mesangial areas, which were characterized by non-branching fibrils arranged in a random array (Figure 2A, 2B). Serum protein electrophoresis showed a decrease in both total protein and total albumin without a monoclonal spike. Urine protein electrophoresis showed an elevated total protein level and an elevated total albumin level. Therefore, the final diagnosis was made of secondary renal amyloidosis due to AOSD.
After one intravenous dosage of 500 mg methylprednisolone pulse therapy was administered. We maintained a high oral daily dose of PD (45 mg/day, 1 mg/kg), CTX (100 mg/day), and COL (1.2 mg/day). Two months later, the PD was tapered as proteinuria decreased. After a follow-up 15 months later, laboratory tests showed that the WBC was 6,000 /uL, Hb 13.2 g/dL, platelet 297,000 / ╬╝L, ESR 11 mm/hr, CRP 0.11 mg/dL, and the serum protein and albumin had increased to 6.2 g/dL and 4.2 g/dL, respectively. The serum ferritin was 24.3 ng/mL. The urine protein clearance was decreased from 6.9 g/day to 92 mg/day. Currently, she is taking PD 7.5 mg, CTX 50 mg, and COL 1.2 gm orally.
DISCUSSION
AOSD is a disorder of unknown etiology and pathogenesis. The association of amyloid deposits and AOSD is rather unusual and should be considered a serious complication. The development of amyloidosis often occurs in patients with longstanding persistent inflammatory diseases5,6). According to previous histopathologic reports on AOSD, complicated renal amyloidosis usually develops after 18 months to 30 years. The incidence of renal amyloidosis in AOSD reveals 4.7% to 14.3%3,4,7ŌĆō12). We have experienced 45 patients with AOSD in our hospital and so far, found one case (2.2%) associated with renal amyloidosis13), which had developed four years after the onset of AOSD.
When this complication occurs, amyloid material is preferentially deposited in the glomeruli, and proteinuria and nephrotic syndrome are the most common initial manifestations. There was no study regarding renal pathology in patients with AOSD. Helin et al.6) demonstrated in a retrospective study of nephropathy in rheumatoid arthritis (RA) that the most common histopathologic finding was mesangial glomerulonephritis (GN), followed by amyloidosis. Amyloidosis was the most common finding in nephrotic syndrome. In a patient with isolated proteinuria, amyloidosis, membranous GN, and mesangial GN were almost equally common. Membranous GN was closely related to gold or D-penicillamine therapies, whereas mesangial GN probably was related to RA itself. Biopsy is thus a valuable tool in differential diagnosis, assessment of prognosis, and decision-making with regard to treatment.
Although either NSAID or an oral steroid is usually effective in AOSD, some patients with complications of the renal amyloidosis may require sustained therapy with corticosteroid and a cytotoxic agent3,5). The COL was also effective in a patient with severe ankylosing spondylitis and nephrotic syndrome due to amyloidosis14). In review of 10 cases with AOSD complicated by renal amyloidosis, combined drug therapy with PD, COL, CTX, azathioprine, and MTX were used3,4,7ŌĆō12). In two of those cases, they were treated with steroids and dialysis due to renal failure4,11). In our experience, it has persistently decreased proteinuria as well as recovered polyarthritis or fever, without any severe complications, during the combined therapy of PD, COL, and CTX (Table 1).
In conclusion, renal amyloidosis must be suspected in patients with AOSD who have unexplained proteinuria. Although the mechanism of renal amyloid deposition is unknown, early diagnosis and treatment with PD, COL, CTX may produce excellent results.

 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print





