


INTRODUCTION
Unlike diseases of the past, which have typically been infections and nutrient deficiencies, the increased average lifespan and excess nutrient supply in modern times have shifted the pattern of diseases and the focus of medicine to aging, cancer, and metabolic diseases. Metabolic conditions, including obesity, metabolic syndrome, and diabetes, are the leading causes of mortality and morbidity. A variety of therapeutic agents and strategies have been developed to combat these conditions. Advances in molecular biology have facilitated the development of therapies targeting specific molecules involved in pathological mechanisms. However, a simple therapeutic strategy comprising restriction of calorie intake and calorie burn is effective against metabolic disorders. In that respect, the importance of lifestyle modification is continuously re-emphasized [1].
Similar to exercise-induced increased calorie burn, increasing cellular energy consumption may also ameliorate metabolic disturbances. Such a therapeutic strategy has potential for diabetic nephropathy (DN) because the hypertrophic response in glomerular cells caused by excess nutrients makes podocytes susceptible to glomerular filtration, resulting in their detachment and loss [2,3]. Adiponectin shows potential in this regard, as it improves insulin sensitivity and increases energy expenditure.
Here we review the characteristics and biological effects of adiponectin and provide our perspective on several issues. We focus on the hypertrophic response in glomerular cells as a pathogenetic mechanism of DN and discuss the role of adiponectin in its attenuation.
ADIPONECTIN AND ITS RECEPTORS
Adiponectin is a 30 kDa peptide adipokine composed of 244 amino acids [4]. It is produced mainly in adipose tissues and reportedly ameliorates metabolic disturbances [5]. It exists in the circulation as various multimers, such as trimers, hexamers, and high-molecular-weight (HMW) forms. The HMW form is predictive of the risk for metabolic syndrome and plays a major role in improving metabolic disturbances. The circulating concentration of adiponectin is high, constituting 0.01% to 0.05% of total plasma proteins [6-9]. Its effects are caused by binding to cognate receptors, including adiponectin receptor-1 (AdipoR1), AdipoR2, and T-cadherin. AdipoR1 is distributed throughout the body, including in glomerular endothelial cells, podocytes, and mesangial and tubular cells in the kidney, but is predominantly in skeletal muscle. By contrast, AdipoR2 is mainly restricted to the liver and its roles are less clear than those of AdipoR1. T-cadherin differs from these in being anchored to the cell membrane and lacking transmembrane structures. In addition, unlike AdipoR1 and AdipoR2, T-cadherin binds only the HMW form of adiponectin. T-cadherin is expressed by endothelial and smooth‑muscle cells and thus is associated with the amelioration of vascular and cardiac conditions [10-12].
DOWNSTREAM SIGNALING PATHWAY OF ADIPONECTIN
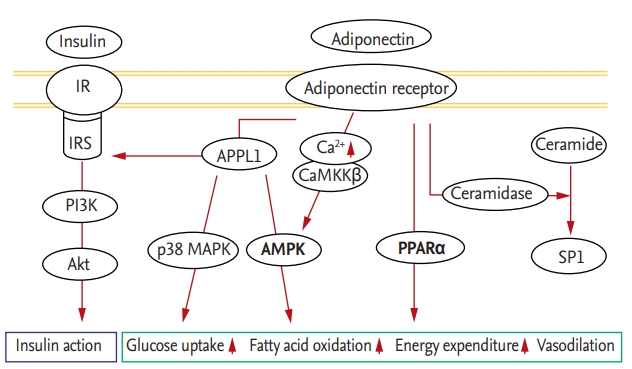
The adiponectin downstream pathway begins with the adaptor protein phosphotyrosine interacting with the PH domain and leucine zipper 1 (APPL1), which binds to the intracellular portion of AdipoR1 and AdipoR2 [13]. When adiponectin binds to AdipoR1, APPL1 activates protein phosphatase 2A, which inactivates protein kinase C-ζ (PKCζ); this in turn dephosphorylates liver kinase B1 (LKB1) in the nucleus, promoting its translocation to the cytosol. In the cytosol, LKB1 activates the 5’-adenosine monophosphate-activated protein kinase (AMPK) pathway, which is responsible for the effects of adiponectin [14,15]. AMPK detects energy deficiency in cells due to depletion or elevation of the intracellular level of ATP or AMP, respectively. Activated AMPK increases cellular uptake of glucose via glucose transporter type 4 (GLUT4), fatty acid oxidation, and vasodilation via endothelial nitric oxide synthase activation to replenish cellular energy [16]. The peroxisome proliferator-activated receptor α (PPARα) pathway is also involved in adiponectin signaling. As a transcription factor, PPARα regulates metabolic processes, and activation of the PPARα pathway promotes intracellular fatty acid oxidation and energy expenditure by upregulating acetyl coenzyme A oxidase and uncoupling proteins [17]. In addition, the insulin pathway is involved in adiponectin signaling. Once insulin and adiponectin bind to their respective receptors, APPL1 directly promotes the interaction of insulin receptor substrate (IRS) with insulin receptors, activating the phosphatidylinositol-3-kinase/Akt pathway and reinforcing the activity of insulin [17]. In addition, APPL1 stimulates glucose uptake via p38 mitogen-activated protein kinase. Adiponectin increases the cytoplasmic Ca2+ concentration by inducing Ca2+ release from the endoplasmic reticulum (ER) or influx of extracellular Ca2+. The resulting elevated Ca2+ level stimulates Ca2+/calmodulin-dependent protein kinase kinase-β, which activates AMPK in a manner independent of APPL1. This activates the NAD-dependent deacetylase sirtuin-1 (Fig. 1) [18,19].
ADIPONECTIN, SOLUTION OF INSULIN RESISTANCE IN ADIPOSE TISSUES
Adiponectin is an insulin-sensitizing hormone [20]. The ability of adiponectin to improve insulin sensitivity has been documented in genetically modified mouse models [21]. To assess the role of adiponectin, an understanding of the functions of insulin and adiponectin, as well as their relationship, is needed (Table 1). Insulin stimulates serum glucose uptake via GLUT4 channels in muscle and adipose tissue and inhibits gluconeogenesis in the liver. In addition, insulin converts glucose into its storage form, glycogen, in the liver and promotes the synthesis of fat in adipose tissue. These effects in combination reduce the serum level of glucose and promote storage of energy. Moreover, insulin has various anabolic effects and is involved in cell proliferation and differentiation [22]. Similarly, adiponectin lowers the serum level of glucose by promoting GLUT4-mediated glucose uptake in muscle and adipose tissues and inhibiting hepatic gluconeogenesis [15,23], and promotes fat synthesis in adipose tissues [24].
However, adiponectin exhibits several metabolic effects that insulin does not. It suppresses free fatty acid synthesis in the liver and induces fatty acid oxidation in muscle [25]. While insulin is an anabolic hormone, adiponectin may be considered a catabolic hormone as it promotes energy expenditure. Although both insulin and adiponectin stimulate cellular uptake of glucose, insulin converts glucose into glycogen or fat for storage whereas adiponectin increases glucose utilization for energy production [26]. In terms of their interaction, insulin and adiponectin stimulate each other [27]. In vitro, insulin increases the expression of the gene encoding adiponectin and the secretion of adiponectin by adipose tissue, although the underlying mechanisms are unclear [28]. Adiponectin has also been reported to increase insulin secretion from pancreatic islet cells in vitro and in vivo [29]. Confusingly, adiponectin seems to have both anabolic and catabolic effects. However, the assumptions below can improve our understanding of the relationship between adiponectin and insulin.
First, adiponectin, an adipokine produced in adipose tissue, promotes adipogenesis. Indeed, it promotes adipocyte differentiation in an autocrine manner [24]; adiponectin-overexpressing mice show morbid obesity and an increased quantity of subcutaneous fat [30]. Furthermore, the preference of adiponectin for subcutaneous fat reduces the quantity of visceral fat and the fat mass of other organs, thereby improving fat tissue quality and insulin sensitivity [30]. Moreover, adiponectin can switch the macrophage phenotype from pro- to anti-inflammatory, which suppresses inflammation in adipose tissue [31,32]. Therefore, adiponectin increases fat synthesis in adipose tissue (an anabolic effect) in contrast to its catabolic effects in non-adipose tissue. Second, insulin is essential for adipocyte growth and maturation [33]. Insulin resistance results in reduced insulin activity in adipocytes despite a high serum level of insulin, and adiponectin rescues adipocytes by reversing insulin resistance. Cytokines, endothelin-1, angiotensin-II, amino acids, saturated fatty acids, and insulin itself contribute to insulin resistance [22]. Interestingly, the metabolic effects of adiponectin are consistent with the reduced levels of these factors in the blood or in tissues. For example, adiponectin lowers the fatty acid level by increasing fatty acid oxidation in muscle and decreasing fatty acid synthesis in the liver. In addition, it lowers serum levels of glucose by increasing cellular uptake of glucose and inhibiting gluconeogenesis in the liver [26]. As a result, it reduces blood levels of insulin, reversing insulin resistance. Adiponectin reduces the cellular level of ceramide, which is implicated in insulin resistance [34]. Independently of AMPK, adiponectin can increase insulin sensitivity by decreasing ceramide levels in the liver and other tissues. In particular, activation of ceramidase by AdipoR1 and AdipoR2 [35] results in conversion of ceramide into sphingosine-1-phosphate, which improves insulin sensitivity [36]. Finally, adiponectin improves insulin sensitivity by interacting with IRS. Above all, the adiponectin-mediated increase in energy release plays an important role in improving insulin sensitivity.
In summary, adiponectin contributes to the survival of adipocytes by maintaining insulin effects on adipocytes, which is mediated by its insulin sensitizing activities in a systemic way.
ADIPONECTIN IN INSULIN DEFICIENCY AND RESISTANCE
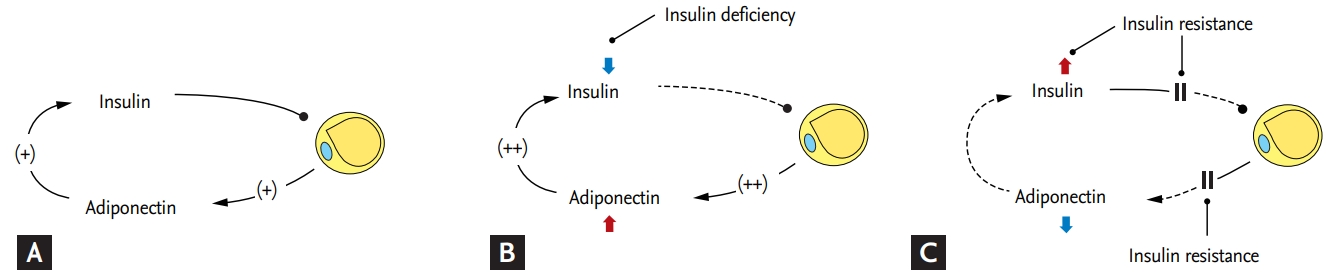
The expression of adiponectin is suppressed in the presence of insulin resistance [37]. The circulating level of adiponectin and the expression of adiponectin receptors (including AdipoR1 and AdipoR2) are reportedly significantly decreased in settings of insulin resistance, e.g., obesity, metabolic syndrome, and type 2 diabetes, compared to under normal conditions [38,39]. This has been hypothesized to be because a low adiponectin level causes insulin resistance and related metabolic disorders. However, because the pathogenetic mechanisms of metabolic disorders are complex and extend beyond adiponectin deficiency, it is more reasonable to presume that a low adiponectin level is primarily due to insulin resistance.
Notably, unlike in patients with type 2 diabetes, the serum level of adiponectin and the expression of its receptors are significantly increased in patients with type 1 diabetes compared to non-diabetic individuals. Patients with genetic defects in insulin receptors also show a significantly increased serum level of adiponectin [40,41]. Thus, adiponectin levels are inversely related to circulating insulin levels. However, these results contradict those from in vitro experiments. Insulin increases adiponectin expression and secretion in 3T3-L1 adipocytes while adiponectin increases insulin secretion in islet cells of the pancreas (Fig. 2A) [28,29]. We suggest that because insulin is crucial for adipogenesis [33,42], the reduced insulin activity caused by insulin deficiency or resistance suppresses adipocyte generation, which results in increased production of adiponectin to maintain insulin activity in adipocytes. As a result, the circulating level of adiponectin is increased by insulin deficiency. This may explain the high serum level of adiponectin in patients with type 1 diabetes and a low insulin level (Fig. 2B). In summary, the high adiponectin level caused by insulin deficiency is an indicator of the insulin requirement of adipocytes. Thus, while a high adiponectin level is predictive of a good prognosis in patients without insulin deficiency, it is associated with poor outcomes in patients with disorders related to insulin deficiency [43].
In the presence of insulin resistance, adiponectin production is reduced by various pathological factors [27], resulting in a low serum level of adiponectin despite the enhanced requirement of adiponectin's role due to a reduced insulin action in adipocytes (Fig. 2C). Because the adiponectin multimer is formed in the ER [44], insulin resistance-induced ER stress activates the unfolded protein response, which suppresses adiponectin synthesis [45]. In addition, obesity-induced inflammation and oxidative stress inhibit adiponectin maturation and secretion [46]. The low serum level of adiponectin induced by insulin resistance reflects metabolic stress and is predictive of a poor outcome in patients with insulin resistance [47,48].
ADIPONECTIN IN KIDNEY DISEASE
Adiponectin is cleared from the circulation in urine and so its serum level is affected by renal clearance. The serum level of adiponectin and AdipoR1/R2 expression in tissue are significantly increased in patients with renal diseases [49]. Specifically, the former is inversely related to the glomerular filtration rate (GFR) [50]. However, reduced clearance of adiponectin does not account for its increased level in patients with renal diseases. Instead, the adiponectin resistance induced by renal diseases leads to elevated adiponectin levels [51]. Indeed, mRNA levels of adiponectin and AdipoR1 are elevated in the adipose tissue of patients with end-stage kidney disease (ESKD) [49]. In addition, interruption at the post-receptor level by uremia causes failure of downstream signaling of AdipoR1/R2, resulting in adiponectin resistance [52]. Therefore, the high serum adiponectin level in patients with chronic kidney diseases, including DN, indicates adiponectin resistance due to renal dysfunction and is predictive of poor long-term outcomes [53,54].
Sharma et al. [55] reported that adiponectin-knockout (APN-/-) mice show normal metabolic profiles when fed normal rodent chow but later exhibit significant albuminuria compared to wild-type mice. Urine testing has revealed oxidative stress with a high level of hydrogen peroxide, and foot-process effacements of podocytes have been observed in renal tissue. However, adiponectin reverses these effects and decreases the magnitude of albuminuria. Furthermore, adiponectin reduces the permeability of hyperglycemic podocyte monolayers to albumin and maintains a normal slit diaphragm structure [55]. In one study, after extensive apoptotic injury in podocytes due to induction of caspase-8 expression, adiponectin-deficient podocyte-specific ablation (POD-ATTAC) mice showed irreversible renal injury that led to ESKD, whereas podocyte recovery was promoted and interstitial fibrosis was reduced in adiponectin-overexpressing POD-ATTAC mice [56]. In addition, APN-/- mice subjected to subtotal (5/6) nephrectomy had a larger glomerular cross-sectional area, a greater number of intraglomerular cells, and more extensive interstitial fibrosis compared to mice subjected to nephrectomy alone. By contrast, in APN-/- mice subjected to nephrectomy in another study, adiponectin reversed all of the abovementioned effects and ameliorated albuminuria [57].
The beneficial effects of adiponectin in the kidney are mediated by its antioxidant, anti-inflammatory, anti-apoptotic, and antifibrotic activities [58]. Notably, the antioxidant capacity of adiponectin plays a crucial role in reducing metabolic injury in patients with DN. Specifically, adiponectin reduces oxidative stress by suppressing the activity of Nox4, an nicotinamide adenine dinucleotide phosphate (NADPH) oxidase abundant in the kidney, via the AMPK pathway [55,59].
UPDATED PATHOPHYSIOLOGY OF DIABETIC NEPHROPATHY
DN is the leading cause of ESKD [60]. Unfortunately, after the development of proteinuria, DN progresses steadily and the GFR declines; this course is rarely reversed. Despite decades-long efforts to identify key pathogenetic mechanisms of DN, the clinical trials performed to date have been fruitless. At present, angiotensin-converting enzyme inhibitors and angiotensin receptor blockers are the only therapeutic agents approved for the treatment of DN [61,62].
The pathophysiology of DN involves renal hemodynamic changes characterized by glomerular hyperfiltration and hypertension, vascular insufficiency with ischemia, oxidative stress with mitochondrial dysfunction, inflammation at the cellular level, and formation of advanced glycation end products. Pro-hypertrophic/fibrotic soluble factors such as insulin‑like growth factor 1, platelet-derived growth factor, epidermal growth factor, vascular endothelial growth factor (VEGF), and transforming growth factor-β (TGF-β) are also implicated. Recent studies have focused on the role of damage or dysregulation at the cellular or genetic level in the pathophysiology of DN. Genetic and epigenetic regulation plays a crucial role in genetic predisposition towards DN. The degree of DNA methylation in DN-related genes contributes to the susceptibility to DN, and microRNAs and histone posttranslational modifications are involved in regulation of the expression of genes related to its pathogenesis [63]. Finally, podocyte autophagy, which is considered a renoprotective process, is interrupted in the presence of diabetic conditions [64].
The pathophysiology of DN is complex and multifactorial, and new findings are continuously being reported. The factors and pathways that play key roles in the pathogenesis of DN are unclear. Nevertheless, glucose excess is implicated in all of these pathogenic mechanisms and remains a hurdle to be overcome.
HYPERTROPHY OF PODOCYTES, GLOMERULI, AND KIDNEY
In nephropathy [65] and DN [2], podocyte injury and loss are the first step leading to glomerulosclerosis. Podocytes are damaged in DN by a variety of pathological mechanisms related to hyperglycemia, including the inflammatory reaction, oxidative stress, induction of apoptosis, dysfunction of autophagy, and glomerular hyperfiltration [66]. Moreover, podocyte loss leads to a decrease in podocyte density, which is predictive of a poor renal outcome.
In general, the hypertrophic response is a result of a compensatory mechanism that maintains functional capacity after glomerulus or podocyte injury [67]. However, glomerular and podocyte hypertrophy plays a crucial role in the pathogenesis of glomerular diseases, including DN [2,3,68]. Thus, below we discuss glomerular and podocyte hypertrophy as a central pathophysiological factor of DN.
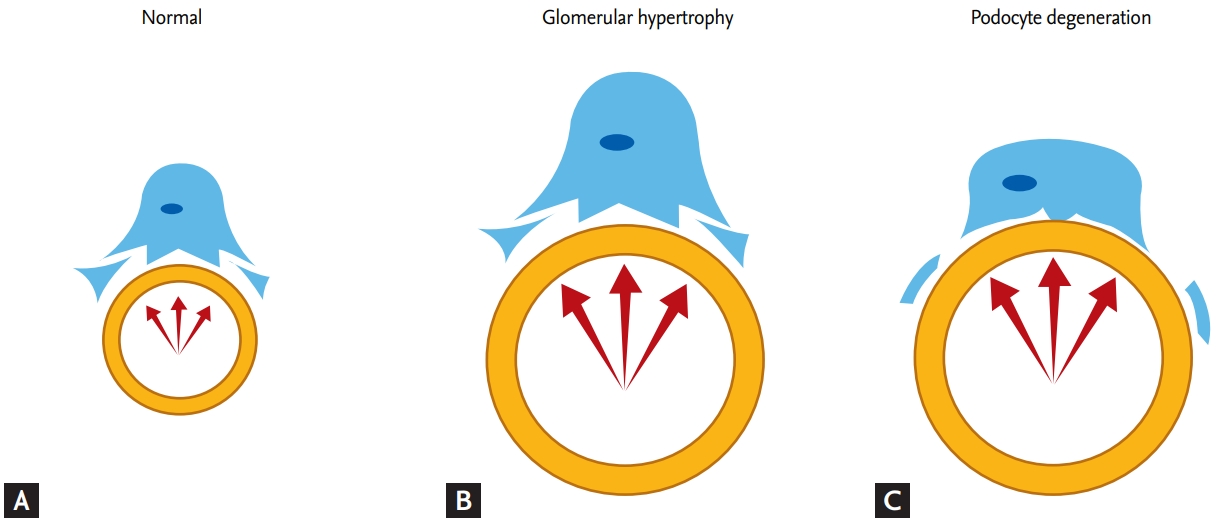
In the pathogenesis of chronic kidney diseases, glomerular loss results in glomerular hyperfiltration and hypertrophy. After renal damage, the surviving glomerulus experiences increased blood flow and initiates hypertrophy at the single-nephron level to compensate for functional loss of other glomeruli [69]. However, the replicative ability of podocytes is limited, and any growth stimulus results in failure of cell division and cytoskeletal reorganization, leading to degradation of the podocyte. Furthermore, the growth of podocytes is principally dependent on cellular hypertrophy, whereas the growth of other cells depends on cellular hyperplasia; thus, the growth of podocytes is less rapid than that of glomerular tufts. As glomerular growth continues, the increasing difference in size leads to podocyte instability due to stretch tension, increasing their vulnerability to the elevated filtration flow caused by the growth of glomerular tufts. Ultimately, glomerular hypertrophy renders podocytes susceptible to detachment from capillaries (Fig. 3) [3,70]. Thus, control of glomerular hypertrophy is crucial for preventing glomerular sclerosis and kidney fibrosis induced by podocyte loss [71,72].
Glomerular hypertrophy is more prominent in DN than in non-diabetic glomerular diseases [68,73]. Moreover, while the hypertrophic response of the glomerulus in non‑diabetic glomerular diseases is an adaptation to glomerular damage at the single-nephron level, glomerulomegaly in DN is extensive and primarily due to continued hyperglycemic stimulation and related growth factors. Renal hypertrophy and elevated renal clearance are observed in early DN but not in other glomerular diseases. But, despite such differences, glomerular hypertrophy is considered as a common process leading to podocyte instability and loss [74]. Thus, suppression of glomerular hypertrophy can prevent podocyte loss in patients with DN [2]. Moreover, the prevention of glomerular hypertrophy is continuous with the prevention of glomerular hyperfiltration. Indeed, renin-angiotensin-aldosterone system (RAAS) blockade, which prevents both glomerular hypertrophy and hyperfiltration, is an effective treatment for DN, supporting the efficacy of therapeutic strategies based on synergy of prevention of glomerular hypertrophy and hyperfiltration.
ADIPONECTIN FOR GLOMERULAR HYPERTROPHY
Glomerular hypertrophy in DN can be prevented by reducing the effects of glucose stimulation on glomerular cells via calorie restriction and calorie burn [1]. Wiggins et al. [75] demonstrated that calorie restriction prevents glomerular and podocyte hypertrophy and results in favorable histologic findings of podocytes and reduced glomerulosclerosis even though a diabetes model was not used in that study. Furthermore, suppression of podocyte growth by podocyte-specific downregulation of mammalian target of rapamycin (mTOR) leads to significant proteinuria and glomerulosclerosis with a mismatch in the size of glomeruli and podocytes [76]. Thus, glomerular hypertrophy can be prevented by controlling the calorie intake, irrespective of the presence of diabetes. However, control of calorie intake by means of lifestyle and nutritional interventions is difficult and its efficacy is inconsistent. Adiponectin represents an alternative treatment option because it reduces blood levels of glucose and improves metabolic disturbance.
Adiponectin decreases cellular hypertrophy; APN-/-mice exhibit greater cardiac hypertrophy because of pressure overload, which is ameliorated by adiponectin [77]. Adiponectin has a similar effect in hyperglycemia-induced cardiac hypertrophy [78]. Indeed, in one study, adiponectin ameliorated kidney hypertrophy in patients with DN. In addition, a specific activator of AMPK, 5-aminoimidazole-4-carboxamide-1-D-ribonucleoside (AICAR), reduced renal hypertrophy induced in mice fed a high‑fat diet and the AMPK pathway played a central role in renal hypertrophy because AICAR had a beneficial effect in mice with adiponectin deficiency [79]. Moreover, APN-/- mice have an increased kidney/body weight ratio compared to wild-type mice, despite their identical blood glucose levels. In addition, Akita mice (a model of type 1 diabetes) with functional loss of adiponectin (Akita/APN-/-) have higher kidney/body weight ratios than healthy counterparts, suggesting that adiponectin negatively regulates the hypertrophic response in the presence of diabetes [80]. Albuminuria indicates glomerular filtration and foot-process effacement, which reflect the state of podocytes. Notably, the albuminuria-reducing effects of adiponectin suggest that it reduces the increase in glomerular filtration and improves the stability of podocytes against the pressure induced by glomerular filtration.
The suppression of glomerular hypertrophy by adiponectin can be explained in various ways. The mTOR pathway is reportedly activated by hyperglycemia, whereas adiponectin inhibits hyperglycemia-induced mTOR activation, thus preventing renal hypertrophy [76]. In addition, adiponectin inhibits the effects of growth factors (e.g., endothelin-1, angiotensin-II, VEGF, and TGF-β) on glomerular hypertrophy [81,82]. A variety of growth factors are involved in the glomerular hypertrophic response to diverse glomerular stresses [74,83]. In a study using an adiponectin-expressing adenoviral vector, adiponectin-treated rats showed reduced glomerular hypertrophy and a significantly lower VEGF level in renal tissue compared to untreated diabetic controls [84].
CURRENT STATE AND FUTURE PERSPECTIVES OF ADIPONECTIN THERAPY
Since its discovery by Scherer et al. [4] in 1995, adiponectin has been shown to have beneficial effects against various diseases. Conceptually, in situations where endogenous adiponectin is reduced by insulin resistance, exogenous administration of adiponectin or an adiponectin receptor agonist is expected to be a good therapeutic strategy. Okada-Iwabu et al. [85] demonstrated the potential of adiponectin for obesity-related diseases, including type 2 diabetes. AdipoRon, an orally active synthetic adiponectin receptor agonist, ameliorates insulin resistance in mice fed a high fat diet and reverses diabetes in db/db mice, which increases their longevity [85-88]. It also ameliorates type 2 diabetes-induced lipotoxicity and oxidative stress, and reverses DN‑specific features in renal tissues [89,90].
Thiazolidinediones, PPARγ agonists used to treat type 2 diabetes, increase plasma levels of adiponectin [91], as does the PPARα agonist fenofibrate [92]. Angiotensin II infusion reduces serum levels of adiponectin, suggesting that the beneficial role of RAAS blockade in renal diseases is due in part to elevated adiponectin levels [93]. Finally, intense exercise increases serum levels of adiponectin [94,95].
Adiponectin shows considerable therapeutic potential but has several drawbacks. First, its overexpression reportedly has adverse effects in animals, such as decreasing bone density, left ventricular hypertrophy, and infertility. Above all, its administration may significantly increase the amount of body fat, despite improvement of systemic insulin sensitivity. Considering that the primary role of adiponectin is to promote adipogenesis, such a paradoxical complication of adiponectin administration is an inevitable consequence. Second, although AdipoRon has an improved safety profile compared to adiponectin, further studies are needed to confirm its clinical efficacy and safety [87].
CONCLUSIONS
The promise of adiponectin therapy may be realized in the near future. Adiponectin has distinctive potential for the treatment of metabolic disorders. The beneficial effects of adiponectin improve principal pathogenetic mechanisms in metabolic disturbance. Moreover, it treats metabolic disorders in fundamental ways as calorie restriction and calorie burn. Thus, adiponectin is a promising therapeutic option for a variety of obesity-related metabolic disorders, including DN.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print





