


INTRODUCTION
Chronic kidney disease (CKD) has become a worldwide health-care concern that calls for prompt initiatives and firm commitments to find effective treatments. The prevalence of CKD is estimated to be 8% to 16% globally and is steadily rising [1]. It not only causes pain in individuals with the disease, but also has effects on society. CKD patients are exposed to an increased risk of death, comorbidities, and cognitive impairment, which all contribute to poor quality of life. Moreover, the medical expenses covering hospitalizations for both cardiovascular and non-cardiovascular events and all-cause mortality are costly. Unfortunately, the end-stage renal disease (ESRD) population is expanding and the CKD population contains potential ESRD patients. Consequently, the expected number of individuals requiring renal replacement therapy (RRT) is tremendous. An example of this prognosis is as follows: if there are currently 100 patients with stage 3 CKD with estimated glomerular filtration rates (eGFRs) lower than 60 mL/min/1.73 m2 10 years from now, 65 of these 100 patients would have already died due to cardiovascular complications, eight would be in need of RRT, and 27 would have ongoing CKD [2]. As the CKD stage deteriorates, there is a consistent increase in the rates of RRT requirement and death. A far greater effect than a doubling of the rates of death and RRT was observed in a 5-year longitudinal follow-up of 27,998 patients, with a mortality and RRT rate of 19.5% and 1.1%, respectively, in patients with stage 2 CKD, and of 45.7% and 19.9%, respectively, in patients with stage 4 CKD [3]. The substantial number of individuals experiencing premature death highlights the urgent need for intervention.
The rise in the prevalence of CKD, along with the subsequent increase in mortality rate, is in part due to the increasing population of the elderly and individuals with diabetes, metabolic syndrome, and cardiovascular disease [4]. As a cause of ESRD, diabetic nephropathy (DN) ranks first both globally and nationwide, with its incidence exponentially increasing from 10% in 1985 to 48% in 2014 according to the Korean ESRD registry [5,6]. DN is clinically characterized by proteinuria (mostly albuminuria), elevated serum creatinine levels, and decreased eGFR. It is well recognized that tight glucose and blood pressure control with renin-angiotensin-aldosterone system (RAAS) blockade constitute the backbone of management strategies for DN. In fact, RAAS blockade has proven to be effective in reducing albuminuria and alleviating the deterioration of DN. Nevertheless, an ever-increasing portion of the DN population is underserved; therefore, seeks novel treatment alternatives. In hopes that treating and deterring the progression of DN may help the prospects of patients with this disease, this review presents recent discoveries and attempts to address some of the unresolved questions that warrant contemplation to provide future direction to overcoming this disease entity.
PATHOGENESIS OF DIABETIC NEPHROPATHY
Hemodynamic and metabolic factors, among which chronic hyperglycemia and dyslipidemia are assumed to play pivotal roles, interact to contribute to the development of DN [7]. The early asymptomatic phase of DN is characterized by an increased glomerular filtration rate (GFR) and renal plasma flow [8]. During hyperglycemia, increased amounts of intraluminal glucose and sodium in the proximal tubule are reabsorbed by upregulation and activation of various co-transporters and exchangers in an effort to achieve euglycemia. Glomerular hyperfiltration results from this hyper-reabsorption in the proximal tubule, which then reduces salt delivery to the macula densa and further decreases blood flow resistance in the afferent arteriole of the glomerulus, contributing to a net increase in single-nephron GFR [9,10]. Hyperglycemia also contributes to the well-recognized pathologic molecular pathways that contribute to renal injury. The production of advanced glycation end products activates polyol and the hexosamine pathways to stimulate the formation of protein kinase C. Mitochondrial overproduction of superoxide exacerbates oxidative stress and promotes inflammation and fibrosis, which induce both functional and structural injuries to the kidney [11,12]. The morphological changes associated with early phase DN comprise thickening of the glomerular basement membrane (GBM), tubular basement membrane and mesangial expansion with characteristic Kimmelstiel-Wilson nodules, along with tubulointerstitial changes and hyalinosis, which result in the replacement of arteriolar smooth muscle cells and the development of sclerotic glomeruli [13].
Dyslipidemia in diabetic patients, especially in type 2 diabetes, is a major reversible risk factor for the progression of renal disease and cardiovascular mortality [14,15]. Interestingly, sustained hyperglycemia in diabetes increases free fatty acid (FFA) synthesis and triglyceride (TG) accumulation in adipose tissue. Further elevation of serum TGs, FFAs, and modified cholesterol levels causes ectopic accumulation of lipids in the parenchymal organs including the pancreas, liver, heart, and kidneys. This process of lipotoxicity serves as an aggravating factor in the pathogenesis of DN in association with glomerulosclerosis and tubulointerstitial injury [16,17]. This lipid byproduct accumulation in DN may arise from altered lipid metabolism resulting from a mismatch between lipid uptake and disposal, featuring enhanced lipid uptake and reduced peroxidation, catabolism, and efflux of residues in the kidney [18].
Systemic hypertension could be both the cause of renal functional deterioration and the consequence of renal damage. Systemically elevated blood pressure due to the co-existence of obesity and cardiovascular disease causes increases in intraglomerular pressure and hyperfiltration, along with subsequent proteinuria [19].
Proteinuria contributes to the development of glomerular hypertrophy and sclerosis by producing local proinflammatory and prosclerotic effects. Both tubular and tubulointerstitial alterations, including hypertrophy, fibrosis, and eventual tubular atrophy, occur along with glomerular changes [13]. In accordance with this, disruption of renal vascular autoregulation by overproduction of vasoactive factors further exacerbates glomerular hyperfiltration. RAAS activation plays a central role by contributing to increased glomerular capillary hydraulic pressure [8]. The production of local angiotensin II increases intraglomerular pressure and proteinuria and stimulates local release of cytokines, further activating inflammatory pathways. This vicious cycle of glomerular hypertrophy and sclerosis, tubulointerstitial inflammation, and fibrosis independently and collectively causes and perpetuates the exacerbation of renal damage [20,21]. Persistent proteinuria followed by a progressive decline in eGFR is characteristic of the late clinical phase of DN, with negatively charged proteoglycan loss in the GBM, podocyte injuries, and malfunctioning tubular cells that ultimately lead to ESRD [13,22]. Furthermore, other factors that potentially contribute to the progression of DN include repetitive episodes of acute kidney injury, comorbid renal and vascular diseases, hyperuricemia, systemic and local inflammation, and tubular cell injury resulting from high glucose exposure.
RENOPROTECTIVE STRATEGIES FOR DIABETIC NEPHROPATHY: THE TRIUMVIRATE
Based on the results of large randomized clinical trials (RCTs), intensive control of glucose and blood pressure might delay the onset, and deter the progression, of DN. Along with this, RAAS blockade constitutes an essential component of the triumviral management strategy for DN. Strict control of both glucose and blood pressure levels can largely influence the rate of eGFR decline. Indeed, albuminuria in patients with DN is associated with a linear decline in GFR, progression to ESRD, and cardiovascular mortality; thus, any means of reducing the extent of proteinuria would be a promising treatment in preventing the progression of DN [23-25].
Deciding whether glycemic control or blood pressure control is of more importance in preventing the progression of DN depends on the clinical circumstances of the individual. During the progression of normoalbuminuria to microalbuminuria, glycemic control plays an important role; blood pressure control; however, does not seem to exert a prominent effect on disease progression. Once microalbuminuria has shifted into a macroalbuminuric state, blood pressure control is of greater importance than glycemic control, in that strict blood pressure restraint helps prevent exacerbation of persistent proteinuria along with renal functional deterioration. However, the significance of glucose control spans the entire disease course, as uncontrolled blood glucose is highly associated with cardiovascular events at any stage of DN. Glycosylated hemoglobin (HbA1c) targets of 6.5% versus 7% and blood pressure targets of less than 125/75 mmHg versus 130/80 mmHg versus 140/80 mmHg are constantly disputed. The Steno-2 study confirmed that a target-driven, long-term, intensified intervention aimed at multiple risk factors (blood pressure < 130/80 mmHg, proteinuria < 0.3 g/day, low density lipoprotein [LDL] < 100 mg/dL, LDL + very low density lipoprotein < 130 mg/dL, HbA1c < 7.0%, smoking cessation, and near normalization of anemia of hemoglobin of 11 to 13 g/dL) in type 2 diabetes patients with microalbuminuria significantly reduced the risks of cardiovascular and microvascular events, including nephropathy, retinopathy, and autonomic neuropathy by approximately 50% [26]. However, there is a growing body of counter-evidence showing that stricter established target levels result in a greater chance of hypoglycemia and cerebrovascular ischemic events in the elderly. Thus, the treatment targets should be tailored according to each individualŌĆÖs clinical situation to optimize renal outcome.
In terms of RAAS inhibition, there have been various attempts to optimize RAAS inactivation with the least number of adverse effects. Several trials have been conducted to validate the efficacy of high dose angiotensin converting enzyme inhibitors (ACEI) or angiotensin II type 1 receptor blockers (ARBs) alone, the combined use of ACEI and ARBs, renin inhibitors (aliskiren), aldosterone inhibitors (spironolactone or eplerenone), and angiotensin II type 2 receptor enhancement. The following studies have reported some promising results. Both the Reduction of Endpoints in NIDDM with the Angiotensin II Antagonist Losartan (RENAAL) trial and the Irbesartan Diabetic Nephropathy Trial (IDNT) showed that ARBs slowed the progression of type 2 DN by slowing the rate of GFR decline compared to placebo groups. This use of ACEIs and ARBs may only provide suboptimal RAAS inhibition and there may still be considerable room for improvement since the normal rate of GFR decline due to aging in those without DN is only 1 mL/min/1.73 m2/year [27,28]. Moreover, a compensatory increase in plasma renin activity as a result of angiotensin blockade led to alterations in angiotensin production and conversion, which suggest the need for direct renin inhibition. Aliskiren is a once-daily, orally effective, small-molecule renin inhibitor that directly targets angiotensin production at its rate-limiting step. It has been proven to produce a non-inferior blood pressure lowering effect relative to that of ARBs and ACEIs with an insignificant side effect profile. ŌĆ£ACE escape,ŌĆØ a phenomenon associated with uncontrolled blood pressure and heart failure, can be avoided with the use of a direct renin inhibitor, which may otherwise occur due to the increased production of angiotensin II with a reactive increase in plasma renin activity through ACE-independent pathways [29-31].
Moreover, aliskiren treatment with losartan proved effective in that it significantly reduced albuminuria and the rate of decline in eGFR, except for relatively infrequent occurrences of hyperkalemia, independently of the baseline CKD stage in patients with DN [32]. A meta-analysis of 11 trials involving 991 patients showed that use of a nonselective aldosterone antagonist along with ACEIs and/or ARBs significantly reduced 24-hour proteinuria and blood pressure in comparison with ACEIs and/or ARBs plus placebo. However, this reduction in albuminuria did not translate into an improvement in GFR and there was a significant increase in the risk of hyperkalemia with the addition of a nonselective aldosterone antagonist to ACEIs and/or ARBs. The long-term effects of these agents on renal outcome, mortality, and safety need to be established [33]. As a last resort for the optimization of RAAS inactivation, dual therapies involving either ACEIs plus ARBs or renin inhibitors plus aldosterone inhibitors were evaluated. Despite these efforts, insignificant effects on renal function as well as increased risks of hyperkalemia and acute kidney injury were demonstrated by both the ALTITUDE (RAS inhibition plus aliskiren) and the VA NEPHRON-D (losartan plus lisinopril) trials.
The effect of statins compared with placebo or no treatment was evaluated in 80 trials with 51,099 individuals. While statins significantly lowered mortality and cardiovascular events in individuals within the early stages of CKD, they had little or no effects in those on dialysis, and had uncertain effects in kidney transplant recipients [34]. Although the mechanism through which statins exert their effects on stroke and kidney function remain to be elucidated, it is certain that statins play an important role in the primary prevention of cardiovascular events and mortality in CKD patients [35].
EMERGING NEW AGENTS IN DIABETIC NEPHROPATHY: BEYOND RAAS INHIBITION
Clinical trials with renal endpoints in patients with diabetes mellitus aim to provide add-on therapies to RAAS blockade. These newer pharmaceuticals are delicately designed to activate or inactivate potentially engaged pathways in the disease course and the narrower spectrums of the drugs are designed to amplify the target effect (Table 1).
Some of these newer drugs for which terminated RCTs have shown negative results include bardoxolone methyl, avosentan, pirfenidone, anti-connective tissue growth factor antibody FG3019, sulodexide, N-acetylcysteine, and ruboxistaurin. Specifically, the Avosentan on Time to Doubling of Serum Creatinine, End Stage Renal Disease or Death in Patients With Type 2 Diabetes Mellitus and Diabetic Nephropathy (ASCEND) trial, which investigated the endothelin receptor antagonist avosentan, was terminated due to an increased risk of fluid overload and consequent congestive heart failure resulting from proximal tubular sodium reabsorption, despite its favorable effect of reducing albuminuria when added to standard treatment [36]. Moreover, dose-dependent peripheral edema is a major barrier limiting their routine use in DN patients. The Ongoing Study of Diabetic Nephropathy With Atrasentan (SONAR) phase III trial with an endothelin A receptor antagonist is approaching its closing date, though some safety issues concerning edema and heart disease are constantly being raised [37]. Bardoxolone, once expected to produce dramatic and powerful antioxidative and anti-inflammatory effects through the activation of the nuclear 1 factor-related factor 2 transcription factor, failed to produce promising results. It was dropped from the Bardoxolone Methyl Treatment: Renal Function in CKD/Type 2 Diabetes (BEAM) and Bardoxolone Methyl Evaluation in Patients with Chronic Kidney Disease and Type 2 Diabetes: the Occurrence of Renal Events (BEACON) trials, owing to the development of various degrees of side effects ranging from trivial muscle cramps to serious issues including heart failure, non-fatal myocardial infarction, stroke, and cardiovascular mortality. It also increased urine albumin excretion and blood pressure [38]. Sulodexide, a highly purified mixture of glycosaminoglycans that potentiates the antiprotease activities of both antithrombin III and heparin cofactor II, had shown beneficial effects in diabetic animal models but failed to prove efficacious in the sulodexide macroalbuminuria (Sun-MACRO) trial [39]. Results from some ongoing RCTs with unestablished renal end points are as follows. Paricalcitol seems effective only at a high dose (2 ╬╝g), and showed only proteinuria lowering effects in the selective vitamin D receptor activation with paricalcitol for reduction of albuminuria in patients with type 2 diabetes (VITAL) study. Evidence regarding its effect in slowing the progression of CKD has yet to be demonstrated [40]. The Proteomic prediction and Renin angiotensin aldosterone system Inhibition prevention Of early diabetic nephRopathy in TYpe 2 diabetic patients with normoalbuminuria (PRIORITY) trial using mineralocorticoid receptor blockers (MRAs; MT-3995, BAY 94-3995, and BAY 94-8862) has shown limited efficacy in the early stages of DN and concerns regarding the development of hyperkalemia need to be addressed [41]. The efficacy of xanthine oxidase inhibitors in terms of delaying the decline in the rate of eGFR was proven in a relatively small sample of CKD patients and will need to be validated in a larger population [42]. The favorable effect of an anti-transforming growth factor ╬▓ (TGF-╬▓) antibody (LY2382770), which was demonstrated in an animal model, has not yet been translated into human data. Pentoxifylline is a methylxanthine derivative and nonspecific phosphodiesterase inhibitor that has anti-inflammatory, antiproliferative, and ant-fibrotic actions. At the end of the Pentoxifylline for Renoprotection in Diabetic Nephropathy (PREDIAN) trial, the rate of eGFR decline and the change in urinary albumin excretion were significantly lower in study participants taking the drug as compared to the placebo group [43]. Stem cell therapy in experimental DN models has not yet shown efficacy in either diabetic mice or rat models [44,45].
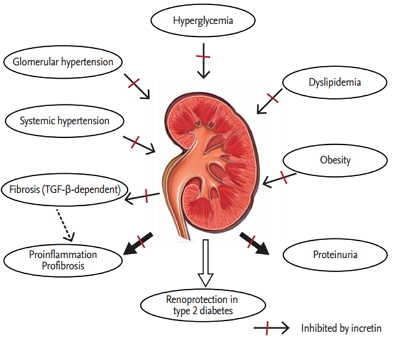
The failure rate of new agents investigated in RCTs exceeds 90%, with barely half of the agents entering phase III trials. These disappointing results may be due to the heterogeneity of the disease entity with its various causes and its diverse underlying pathogenic mechanisms that may be intertwined. Moreover, the flexible inclusion criteria of the trials, including variable stages of DN ranging from relatively spared renal function to advanced CKD, may make it difficult to infer causality. Although changes in proteinuria levels are frequently used as a parameter in predicting renal outcome, proteinuria per se cannot be translated into eGFR; thus, other consolidated surrogate endpoints representing renal outcomes are needed (Fig. 1). Despite the inconsistent and questionable results of the trials involving newly developed agents, the following agents have been proven to demonstrate somewhat favorable renoprotective effects through inhibiting intrinsic renal pathways linked to inflammation and fibrosis.
Incretin-based agents
Incretins are gut hormones that are secreted into the circulation postprandially to exert insulinotropic activity. Glucose-dependent insulinotropic polypeptide (GIP) and glucagon-like peptide 1 (GLP-1) are the most important incretins released in humans, as their potential use for the treatment of diabetes appears promising. Impaired incretin regulation is implied in type 2 diabetes based on findings that meal-induced increases in GLP-1 levels were reduced, whereas the level of GIP was retained in the normal range in subjects with diabetes [46]. Thus, defective GLP-1 release may be responsible for the characteristic metabolic disarray presented in type 2 diabetes. Although the mechanism involved in defective GLP-1 secretion has been unraveled, the preservation of the GLP-1 response to oral glucose in those with prediabetes suggests that its impaired release may be a consequence of the diabetic condition rather than the primary incident [47]. Moreover, the insulin response to exogenously administered GIP is reduced in type 2 diabetes [48]. This impaired secretion of GLP-1 and the defective insulinotropic action of GIP constitute potential therapeutic targets in subjects with type 2 diabetes. The potential use of these insulinotropic gut peptides has been most successful for GLP-1, which exerts antidiabetogenic properties by stimulating insulin secretion, increasing ╬▓-cell mass, inhibiting glucagon secretion, delaying gastric emptying, and inducing satiety [49-51]. Despite these favorable results with GLP-1, its rapid degradation by dipeptidyl peptidase 4 (DPP-4) makes it somewhat impractical. However, this limitation can be overcome using DPP-4 resistant GLP-1 receptor agonists and DPP-4 inhibitors. Apart from their renowned glycemic effects of improving insulin secretion and inhibiting glucagon secretion, both GLP-1 receptor agonists and DPP-4 inhibitors are known to reduce blood pressure, dyslipidemia, and inflammation to a certain degree with low risk of hypoglycemia (Fig. 2).
With respect to their renal effects, these incretin-based agents have been shown to reduce albuminuria by inhibiting renal tubular sodium reabsorption and subsequent increases in glomerular pressure in both rodents and humans. Furthermore, the onset of the morphological alterations associated with DN were delayed or prevented with these drugs in rodent models [52]. There are currently a few results from phase III trials suggesting the potential therapeutic effects of sitagliptin, alogliptin, and linagliptin in significantly reducing the amount of albuminuria in patients with type 2 diabetes mellitus [53-55]. Treatment with exendin-4 or exenatide has been shown to be associated with reductions in both albuminuria and urinary levels of TGF-╬▓ and type IV collagen in the same population [56]. Although it is well recognized that renal fibrosis is primarily mediated through TGF-╬▓, there is limited clinical evidence of the effect of targeting TGF-╬▓. The anti-fibrotic effect of linagliptin was demonstrated in a type 1 model of DN after 4 weeks of treatment. The mitigation of fibrotic changes due to DN was associated with an inhibition of endothelial-to-mesenchymal transition and the restoration of microRNA 29s [57]. In human renal proximal tubular cells, linagliptin seems to interfere with the activation of TGF-╬▓. Moreover, the cation-independent mannose 6-phosphate receptor (CIM6PR) is crucial for the conversion of latent to active TGF-╬▓1 in human kidney proximal tubular cells [58]. The co-existence of DPP-4 and CIM6PR on the cell membrane is highly suggestive of the reduced activation of TGF-╬▓, through which linagliptin may have exerted its TGF-╬▓1-dependent antifibrotic effects (Fig. 2) [59]. From a clinical standpoint, this finding is relevant because of the potential of linagliptin as an antifibrotic agent in patients with diabetes.
Sodium/glucose co-transporter 2 inhibitors
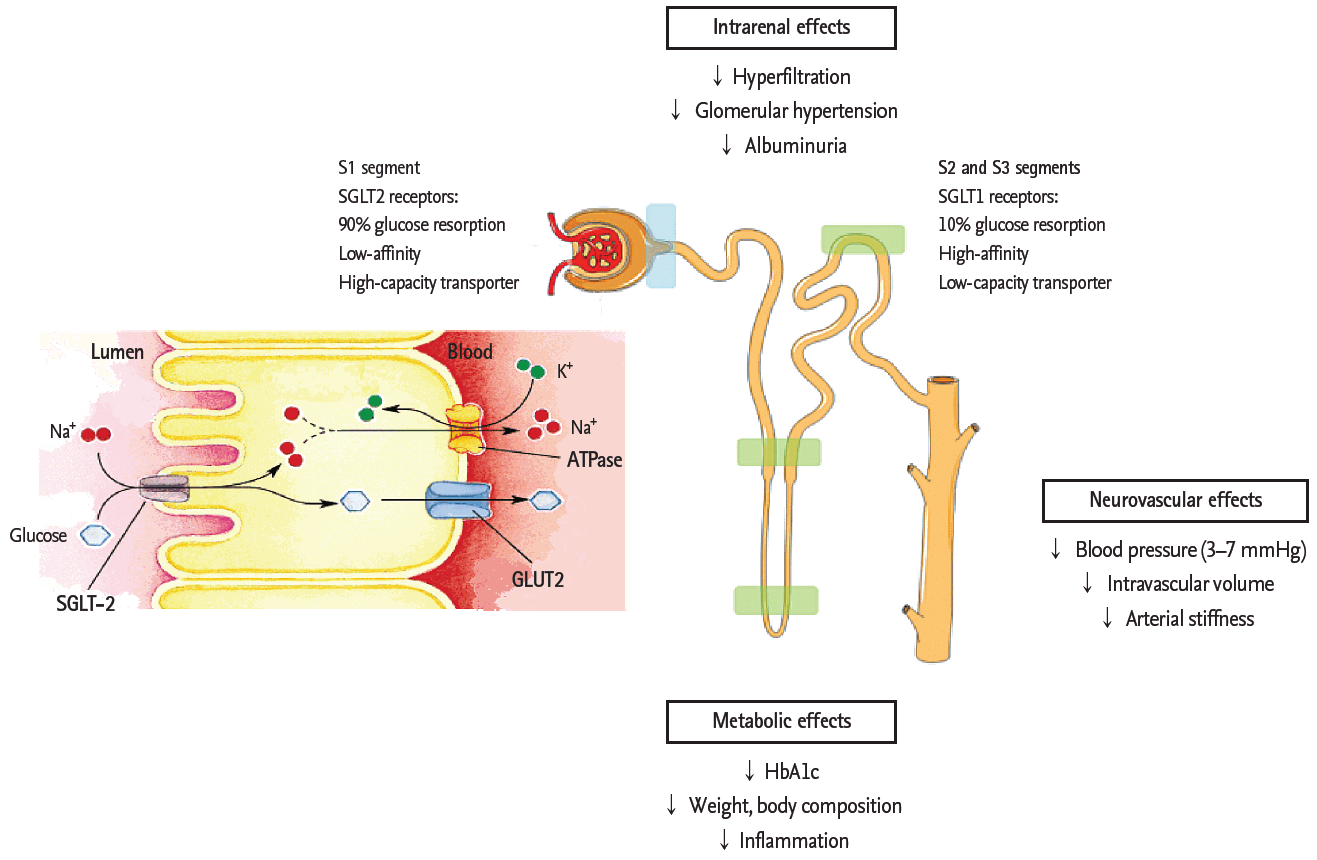
Sodium/glucose co-transporter 2 (SGLT2) inhibitors are promising hypoglycemic agents that have the added advantage of not promoting hyperinsulinemia, weight gain, or hypoglycemia, unlike traditional antidiabetic agents [60]. Their mode of action lies in the effective mediation of glucose reabsorption in the kidney by blocking glucose entry into the renal proximal tubular cell; thereby, causing glucose excretion via urine. Decreased proximal tubular glucose reabsorption with SGLT2 inhibition distributes the burden of salt balance in the early proximal tubule to the distal segments of nephrons. At the same time, increased sodium delivery to the macula densa restores tubuloglomerular feedback, which modulates the tone of glomerular afferent resistance through the release of the vasoconstrictive substance adenosine, which further mitigates glomerular hyperfiltration (Fig. 3) [61]. This net reduction in glomerular hyperfiltration helps break the vicious circle that is implicated in the early phase of DN. Moreover, the renoprotective effect of SGLT2 inhibitors, particularly empagliflozin, has been demonstrated through a decrease in high glucose-induced inflammatory and fibrotic markers in human proximal tubular cells. High glucose-induced expression of Toll-like receptor-4, binding of nuclear deoxyribonucleic acid to nuclear factor ╬║B and activator protein 1, and secretion of collagen IV and interleukin-6 were attenuated with empagliflozin; these effects were most likely due to blocking glucose entry into the cell [62].
Several trials have evaluated the potential renoprotective effect of SGLT2 inhibitors in large cohorts of diabetic patients. In one trial, patients treated with canagliflozin had a decrease in the urinary albumin-creatinine ratio and a slight decrease in eGFR [63]. The slight decrease in eGFR required a dose adjustment in those with renal impairment. On the other hand, this calls into question the reliability of GFR as a trusted variable, especially in those with early phase diabetes with hyperfiltrating nephrons. Moreover, drug dose-dependent hyperkalemia and urogenital infections were reported from time to time with negligible clinical impact [64]. In the most recent trial involving diabetic patients with moderately decreased renal function, the dapagliflozin group showed favorable results in terms of reduced albuminuria [65]. Along with this, favorable metabolic effects, including a reduction in HbA1c levels, decreases in body weight and blood pressure due to osmotic diuresis, and a loss of calories related to glycosuria, indicate the potential therapeutic effect of SGLT2 inhibition in DN (Fig. 3) [66,67].
5ŌĆÖ Adenosine monophosphate-activated protein kinase activators
An imbalance between energy intake and expenditure gives rise to alterations in glucose and lipid metabolism that eventually lead to the development of fasting and postprandial hyperglycemia, together with dyslipidemia and insulin resistance, which constitute the characteristic features of diabetes [68]. Chronic exposure to glucose overload, free fatty acids, and amino acids exerts toxic effects in various relevant organs [69]. In this respect, DN is the end result of multifactorial processes that stem partly from this glucolipotoxicity. A 5ŌĆÖ adenosine monophosphate-activated protein kinase (AMPK) is a metabolic master switch that regulates downstream signals based on shifts in surrounding energy reservoirs [70]. There is mounting evidence demonstrating that the dysregulation of AMPK in relevant tissues is implicated in the development of metabolic syndrome and diabetes [70]. As AMPK activation is proven to improve glucose and lipid homeostasis in insulin-resistant animal models by coordinating anabolic processes, targeting this enzyme may ameliorate some of the pathologic features of DN [71,72].
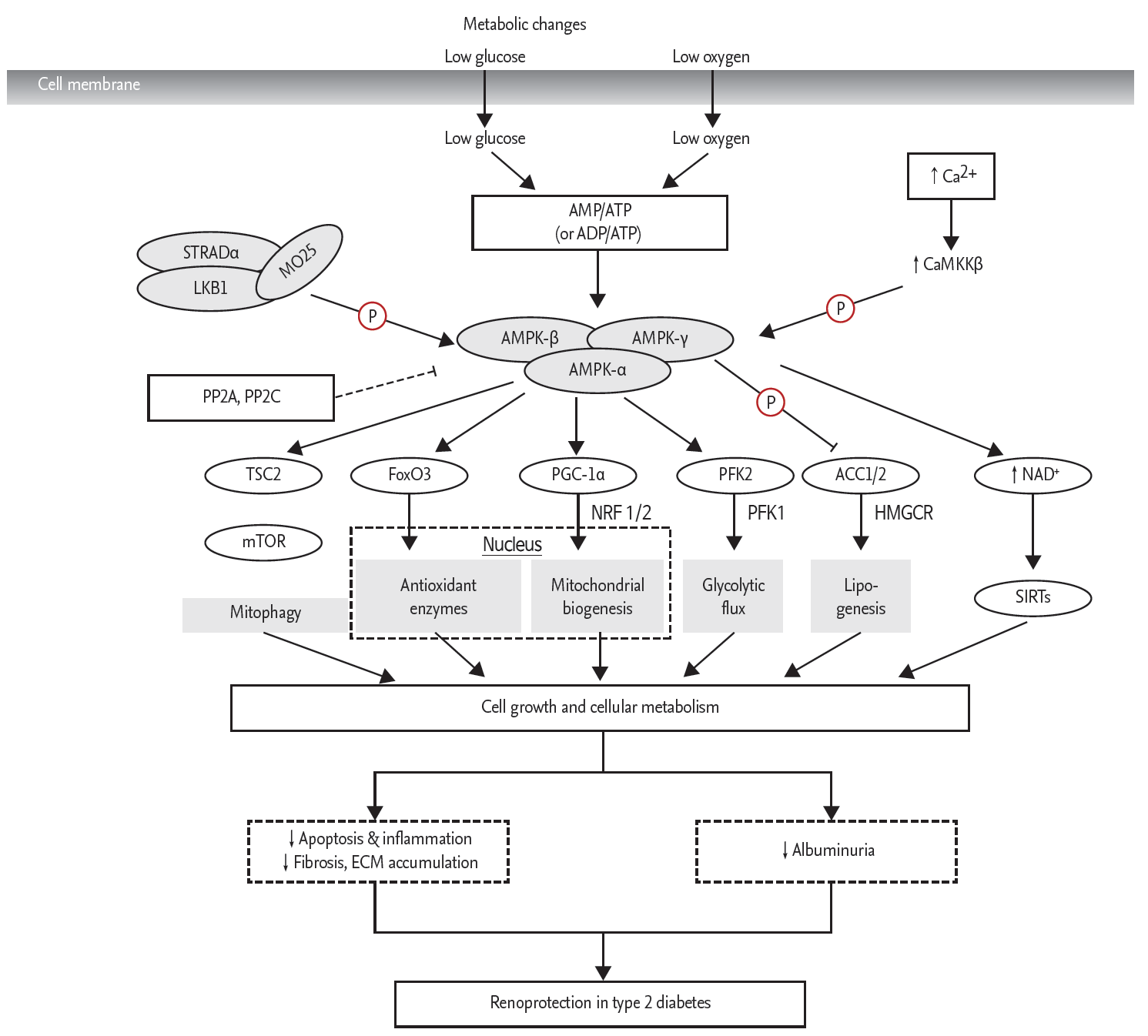
AMPK is activated during metabolic stress in which adenosine triphosphate (ATP) consumption causes an increase in the adenosine monophosphate (AMP)/ATP ratio [70]. It is also stimulated by several upstream kinases, including a compound molecule that consists of three proteins: STE-related adaptor (STRAD), mouse protein 25 (MO25), and the tumor-suppressor liver kinase B1 (LKB1). Ca2+/calmodulin-dependent protein kinase kinase ╬▓ (CaMKK╬▓) and TGF ╬▓-activated kinase (TAK1) are upstream enzymes that participate in the cellular signaling cascade of AMPK activation [73,74]. In response to oxidative stress, activated AMPK phosphorylates its main downstream targets, acetyl-CoA carboxylase and hydroxymethylglutaryl CoA reductase. These are primarily involved in the rate-limiting steps of lipid homeostasis, which further promote fatty acid oxidation upon phosphorylation by AMPK [75]. AMPK activation triggers several adaptive responses for cell survival, which are induced by enhanced oxidative stress due to mitochondrial dysfunction. These downstream targets of AMPK include tuberous sclerosis complex 2, peroxisome proliferator-activated receptor ╬│ (PPAR╬│) coactivator-1╬▒ (PGC-1╬▒), and Forkhead box O3 (FOXO3), which, upon phosphorylation by AMPK, fortify cellular autophagy, antioxidant defense, and mitochondrial biogenesis through the inhibition of the mammalian target of rapamycin complex 1 in affected cells (Fig. 4) [76]. The sirtuin 1 (Sirt1) protein is the founding member of a family of NAD+-dependent deacetylases and is linked to longevity associated with calorie reduction. Sirt1 activity is regulated via the availability of its substrate, NAD+ [77]. In type 2 diabetic animal models [72,78] and human kidneys [79], the expression and activity of Sirt1 was significantly reduced. The mechanism of Sirt1 reduction in the diabetic condition has not been clarified, but a reduction in the phosphorylation of AMPK may play a role [72]. This notion has been supported by recent publications, which showed that AMPK activation by resveratrol and theobromine leads to Sirt1 activation, which protects the diabetic kidney (Fig. 4) [72].
Conventional activators of AMPK include 5-aminoimidazole-4-carboxamide ribonucleoside, metformin, adiponectin, and resveratrol, which have been shown to provide additional renoprotective effects in addition to their intrinsic activities [72]. Some of the recently recognized AMPK activators are under investigation. Fenofibrate-fed diabetic mice demonstrated both renal functional and phenotypic improvements through reduced lipotoxicity via stimulation of the AMPK-PGC-1╬▒-ERR-1╬▒-FOXO3a signaling pathway [77]. Anthocyanin, a flavonoid in the polyphenol class, has also been shown to attenuate lipotoxicity-related apoptosis and oxidative stress in DN through the activation of AMPK and its downstream effectors [78]. Moreover, our unpublished data suggest cinacalcet as a potential therapeutic agent in ameliorating pathologic alterations in DN. Cinacalcet is a calcimimetic that has been shown to increase intracellular Ca2+ and positively modulate the calcium-sensing receptor and subsequently activate the CaMKK╬▓-LKB1-AMPK signaling pathway in the kidney. AdipoRon, an adiponectin receptor agonist, is an orally active, synthetic molecule that has demonstrated favorable effects in DN through the activation of the AMPK and PPAR╬▒ pathways via up-regulation of the adiponectin receptor 1 and 2 in our preliminary data. Taken together, there have been constant attempts to verify the favorable renoprotective effects of AMPK activation in diabetic rodent models that have yet to be translated into human data.
CONCLUSIONS
In response to the constantly growing burden of diabetes and its associated complications including DN, research into novel therapies to treat DN is expanding. Currently, the management strategy for DN consists mainly of antihypertensive and antiproteinuric measures that depend on strict RAAS inactivation. However, these traditional therapies have been suboptimal and there is a clear, unmet need for treatments that offer effective schemes beyond glucose control. The complexity and heterogeneity of the disease entity, along with the ambiguous renal endpoints, which deter accurate appraisal of new drug potency, are making the situation worse. In fact, albuminuria and eGFR do not appear to be reliable variables that reflect the true renal function of each individual with different stages of DN (Fig. 1). Thus, research into original therapies to treat DN is focusing on the intrinsic renal pathways that intervene with intracellular signaling of both anti-inflammatory and antifibrotic pathways. Mounting evidence in support of the favorable metabolic effects of these novel agents with respect to the renal aspects of DN supports the likelihood of systemic beneficial effects as well. Thus, when translated into clinical use, these novel agents would also address the comorbid factors associated with diabetes, such as obesity and risk of cardiovascular disease.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print





