


INTRODUCTION
Shwachmann-Diamond syndrome (SDS) is a rare autosomal recessive disorder characterized by exocrine pancreatic insufficiency, somatic abnormalities (particularly metaphyseal dystosis) and bone marrow dysfunction1,2). It was first described in 1964 by Shwachman et al. and is now recognized as the second most frequent cause of pancreatic insufficiency in children1). In addition, myelodysplastic syndrome and acute myeloid leukemia (AML) have been recognized as major complications in this disease and occur in up to one third of patients3). When leukemia develops, due to the preceding bone marrow failure, serious complications may be induced by chemotherapy and underlying organ dysfunctions. At present, the only curative treatment option for bone marrow dysfunction and leukemia in SDS is allogeneic bone marrow transplantation (BMT)4,5). These patients should be managed with extreme caution when treated with allogeneic BMT.
We report the first case in Korea of a patient with SDS to undergo related BMT following transformation to AML.
CASE REPORT
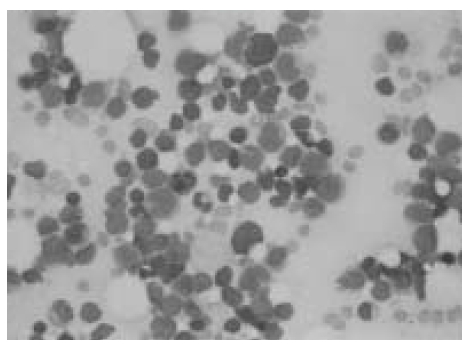
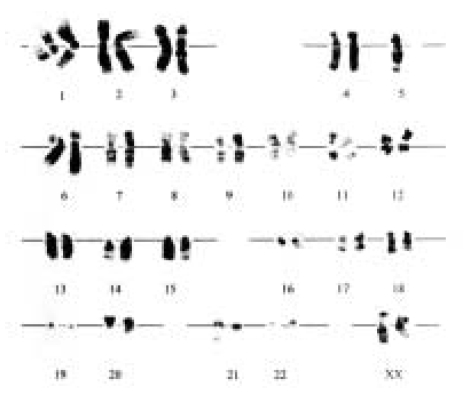
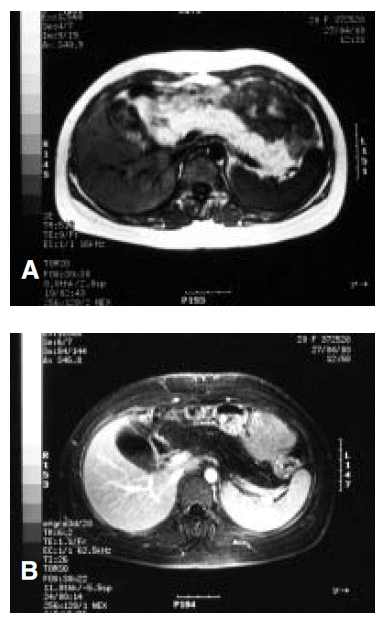
A 21-year-old woman was admitted to the hospital because of indigestion and poor oral intake. Her most prominent clinical features were short stature, diarrhea and hematologic abnormality. She was pancytopenic (Hb 9.7 g/dL white cell count of 4,200/╬╝L [neutrophils 800/╬╝L] and platelets 220,000/╬╝L). Marrow aspirate showed high cellularity with massive infiltration by blast cells which were positive for Sudan black and myeloperoxidase (Figure 1). She was diagnosed as AML, M2 according to the FAB classification. The blast population displayed the following immunophenotype: CD13 48%, CD33 59%, CD34 61%, HLA-DR 59% and CD20 negative. A bone marrow sample was processed for cytogenetic analysis. A complex karyotype was found in 20 out of 20 metaphases: 47XX, del (20)(q11.2), tas (21;22) (q22;q13), +22 (Figure 2). A skeletal survey showed an mild metaphyseal dystosis. The abdominal MRI showed an enlarged pancreas, a hyperintense signal on T1-and T2-weighted image and low signal on fat-suppressed image (Figure 3A, B). She was treated with conventional chemotherapy (idarubicin plus cytarabine). The period of aplasia was characterized by numerous complications, including persistent fever despite empiric antibacterial and antifungal therapy and hematemesis. Marrow recovery was slow. The patient achieved remission 41 days after the beginning of chemotherapy. Conventional cytogenetics at that time showed a normal karyotype. She received BMT from her sister who was HLA identical. Unmanipulated donor marrow was given following conditioning with cyclophosphamide (60 mg/kg├Ś2) and total body irradiation (TBI) (1200 cGy in 6 fractions) using a combination of cyclosporin A and methotrexate as graft-versus-host disease (GVHD) prophylaxis. The post-transplant period was complicated by staphylococcal septicemia and acute GvHD in grade II. But she has been alive disease free and off any immunosuppressives, for a follow-up of 13 months after BMT.
DISCUSSION
SDS is an inherited constitutional bone marrow failure syndrome and has a marked propensity of developing leukemia5). In a recent study, the overall risk of malignant transformation was found to be 24%, while leukemia was observed in 70% of a subgroup of patients with pre-existing pancytopenia, myelodysplastic features and clonal cytogenetic abnormalities2,6).
Previous to this report, BMT has been documented in some patients with SDS7). One patient died from congestive heart failure attributed to cyclophosphamide toxicity as part of the conditioning regimen8). Although a similar cyclophosphamide dose was used in the patient reported here, no cardiac dysfunction was observed in the pre- and post-BMT period. Our observations indicate that a cyclosphosphamide/TBI conditioning regimen might be taken into consideration in patients with SDS.
The aetiopathogenesis of SDS is still enigmatic. The unspecific cytogenetic abnormalities are a common finding in SDS and were detected in about 40% of patients in an earlier study2). This led to the hypothesis that SDS might also be a chromosomal breakage syndrome caused by a DNA repair defect9). And also, constitutional karyotype of our patient was abnormal: 47XX, del (20) (q11.2), tas (21;22) (q22;q13), +22.
As the same with other cases of acute leukemia in patients with SDS, the clinical course of the disease in our patient was characterized by multiple complications after chemotherapy, and by a delayed hematologic reconstitution2,10). Experiences with allogeneic BMT are extremely limited. Although the correct clinical approach for leukemic SDS patients is still to be determined, our case shows that a stable remission may be obtained by allogeneic BMT, even in the presence of an adverse complex bone marrow karyotype.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print





