


INTRODUCTION
Cardiomyocytes in the fetal development exhibit abundant proliferative capability, whereas cardiomyocytes in the adult mammals exhibit little, if any, capability to undergo cell division1ŌĆō4). Consequently, myocardial loss due to injury or disease during adulthood is irreversible, and the potential therapeutic value for myocardial regeneration is quite substantial. Knowledge of the mechanisms that control the cardiomyocyte cell cycle would allow us to design procedures to initiate repair or regeneration of the adult myocardium following injury. Unfortunately, we have yet to determine how the mammalian cardiomyocyte cell cycle is blocked after birth.
A number of cell-cycle regulated proteins have been identified and categorized as either cyclins, cyclin dependent kinases (CDKs) or cyclin dependent kinase inhibitors (CKIs)5ŌĆō7). The activity of CDKs control cell cycle progression, which activity is regulated positively and negatively by the association of CDKs with cyclins and CKIs, respectively8, 9). Cyclins and CDKs are classified as G1 (gap1), S (synthetic) or G2 (gap2)/M (mitotic) phase proteins, depending on the role and protein abundance during the cell cycle5ŌĆō7). Presently, two families (INK and CIP/KIP) of CKIs exist, and include p15INK4B/p16INK4A/p18INK4C/p19INK4D and p21C IP 1/p27KIP 1/p57KIP2 10, 11). While the former family of CKIs bind to and inhibit only G1 phase cyclin-CDK complexes, the latter family of CKIs bind to and inhibit G1 and S phase cyclin-CDK complexes which leads to G1/G0 cell cycle arrest10, 11).
Recently, the importance of cell-cycle regulated proteins in cardiomyocytes has been recognized12ŌĆō16). Yoshizumi et al.12) reported that the disappearance of cyclin A, which is involved in DNA synthesis, correlates with permanent withdrawal of cardiomyocytes from the cell cycle in human and rat hearts. We have reported the reduction of various cyclins and CDKs protein expressions with differential and dramatic decrease of CDK activities in cardiomyocytes during the neonatal period, and we have hypothesized that the functionally active cdc2 in neonatal cardiomyocytes is involved in binucleation formation16).
However, CDK activities and expression of CKIs in human hearts during development have not been examined. Therefore, we examined the expression and activities of cyclins, CDKs and CKIs in human atria during development in the human. We found that marked reduction of protein levels and activities of cyclins and CDKs, and marked induction of p27KIP 1, are associated with the withdrawal of cardiac cell cycle in adult humans.
MATERIALS AND METHODS
Sampling
Since the sampling of fresh ventricular tissues is very difficult from adult humans, we have chosen the sampling of atrial tissue for this study. Atrial tissues of adult human were obtained from patients (48ŌĆō55 years old) with mitral valvular disease during open heart surgery. Atrial tissues of fetus were obtained from gestation week 12 and 25-old fetus whose mother underwent surgery due to threatened abortion or premature labor. All samplings were conducted in our hospital from July 1997 to November 1997. Informed consent was obtained from each patient. Atrial tissues were immediately frozen in liquid nitrogen and stored 70┬░C until assayed, while the remaining half of specimens were fixed in 10% neutral formalin and routinely processed for pathologic study. Sprague-Dawley hosts and pregnant females were obtained from our breeding colony. Onset of pregnancy in female rats was determined by vaginal plug. Pregnant rats were killed at day 17 by decapitation and embryos were removed, decapitated and ventricles were harvested and stored in liquid nitrogen. Hearts were removed and ventricles were collected in the same manner from adult animals.
Western blot
Samples were homogenized in Nonidet P-40 buffer containing several protease inhibitors. Protein was quantitated, then separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) system and electrotransferred to nitrocellulose membranes16). The nitrocellulose membranes were blocked by incubation in blocking buffer, incubated with primary antibody, washed, incubated with horseradish peroxidase conjugated secondary antibody, and signals were visualized by the ECL detection method according to the manufacturerŌĆÖs protocol (Amersham Int. plc, Buckinghamshire, UK). Anti-cyclin D1 monoclonal antibody (72-13G), anti-cyclin D3 polyclonal antibody (C-16), anti-CDK4 polyclonal antibody (C22), anti-CDK2 polyclonal antibody (M2), anti-cdc2 polyclonal antibody (pSTAIRE), anti-PCNA monoclonal antibody (PC10), anti-p21C IP 1 (C-19) and anti-p27KIP 1 (N-20) were purchased from Santa Cruz Biotechnology Inc. (Santa Cruz, CA, USA). Anti-cyclin A and anti-cyclin B monoclonal antibodies were generously provided by Dr. Michael Brandeis (ICRF Clare Hall Lab. Cell Cycle Control, UK).
Immunoprecipitation and assay of each CDK activity
To measure CDK4 or CDK2 activity, the method of Jahn et al.17) was slightly modified16). Protein lysates (500 g) were diluted with 1 ml (final volume) of RIPA-buffer [1% Triton X-100, 1% sodium deoxycholate, 0.1% sodium dodecyl sulfate (SDS), 0.15 M NaCl and 0.01 M Tris, pH 7.4] and precleaned with 50 ╬╝l of protein A-Sepharose CL-4B beads (0.15 g/ml, Pharmacia Biotech, Uppsala, Sweden) for 1 h at 4┬░C. The beads were then removed by centrifugation. Two g of CDK antibody was added to precleaned lysates in 1 ml of RIPA-buffer, and the mixture was gently rocked for 2 h at 4┬░C. Thirty ╬╝l of protein A-Sepharose CL-4B beads were added and the mixture was gently rocked for an additional 1 h at 4┬░C. Immunocomplexes were collected by centrifugation (3,000g, 3 min, 4┬░C) and washed three times with ice-cold RIPA-buffer. Half of the immunocomplex bound beads was eluted in sample buffer, separated by SDS-PAGE, and Western blot analysis was performed as described previously. Purified CDK kinase activity in immunoprecipitates was measured using a modification of the methods previously described16). Products of retinoblastoma protein (pRb, Santa Cruz) was used for determining the activities of CDK4 or CDK6 as the substrate. Histone H1 (Calbiochem, La Jolla, CA, USA) was used for determining the activities of CDK2 or cdc2 as the substrate. The remaining half of immunocomplexes bound beads was suspended in kinase buffer (10 mM MgCl2, 5 mM MnCl2, 1 mM DTT, 50 mM HEPES-NaOH, pH 7.3) and washed twice in kinase buffer. Reactions were carried out in kinase buffer (final volume, 50 ╬╝l) with 0.1 ╬╝g of pRb or 2.5 ╬╝g of histone H1, 0.1 mM ATP and 5.0 ╬╝Ci [╬│-32P]ATP at 30┬░C for 20 minutes and stopped by adding 2├Ś SDS sample buffer. The optimum incubation time was determined by measuring several time points of pRb or histone H1 phosphorylation (data not shown). Final reactants were separated using SDS-PAGE, fixed with 5 to 10 volumes of glacial acetic acid:methanol:water (10:20:70), dried and autoradiographed. Signals were quantified using densitometry.
RESULTS
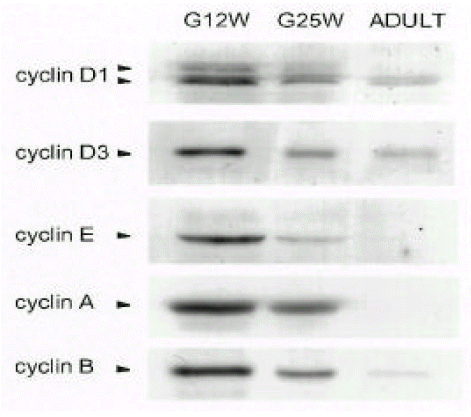
Temporal changes of cyclin protein levels in human atria during the fetal and adult period (Fig. 1)
Anti-cyclin D1 antibody recognized two cyclin D1 bands that indicated phosphorylation status. The two cyclin D1 bands were readily detectable at gestation 12 weeks (G12W) and gestation 25 weeks (G25W). As the phosphorylated active from cyclin D1 (upper band) disappeared, the dephosphorylated inactive from cyclin D1 (lower band) was detectable at the adult period. The cyclin D3 levels decreased gradually from the early fetal period to the adult period. Cyclin E, A and B levels were high at G12W, decreased gradually at G25W and were not detectable at the adult period.
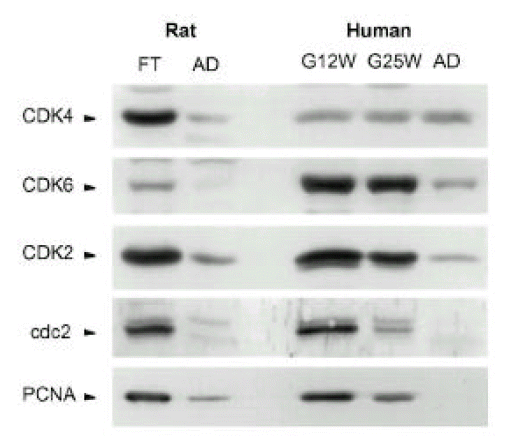
Temporal changes of CDK protein levels in rat ventricles and human atria during the fetal and adult period (Fig. 2)
The protein levels of CDKs and PCNA were high at the fetal period and their levels decreased markedly at the adult period in the rat. The CDK4 and CDK6 levels were high at G12W in human atria, and those did not change during the fetal period. Interestingly, while CDK4 levels did not change, CDK6 levels decreased from the fetal period to the adult period. CDK2, cdc2 and PCNA levels were high at G12W, decreased gradually at G25W and decreased markedly at the adult period.
Changes of CDK activities in human atria during fetal and adult period (Fig. 3)
The specific antibodies for CDKs that were used for Western blot analysis were used to purify and measure the functional activities of CDKs. Each antibody bound its respective protein in atrial lysates, and these immune complexes were immunoprecipitated as confirmed by Western blotting (data not shown). While CDK4 activity did not change from the fetal period to the adult period, activities of CDK6, CDK2 and cdc2 decreased markedly. The ratio (adult period to fetal period) of each CDK activity was 0.89 0.11 (CDK4, n=3, not significant), 0.12 0.02(CDK6, n=3, p<0.05), 0.18 0.04 (CDK2, n=3, p<0.05, CDK2) and 0.09 0.02 (cdc2, n=3, p<0.05)
Temporal changes of p21C IP 1 and p27KIP 1 protein levels in human atria during fetal and adult period (Fig. 2)
The protein levels of p21C IP 1 and p27KIP 1 were readily detectable in atria during the fetal period. Interestingly, while p21C IP 1 in atria was not detectable at the adult period, the protein levels of p27KIP 1 increased (5.6┬▒1.2 fold, n=3, p,0.05) significantly in the adult period.
DISCUSSION
The present study is designed to determine the changes of cyclins, CDKs and CKI proteins in human atria during the fetal and adult periods. These findings indicated that the protein levels of most cyclins and CDKs were high in atria during the fetal period, then they decreased at different rates. While the activities of CDK6, CDK2 and cdc2 decreased markedly, the activity of CDK4 was not changed. While p21C IP 1 protein in atria was detectable only in the fetal period, p27KIP 1 protein increased markedly in the adult period. In this study, cardiomyocytes may have contributed greatly to the levels of cyclin, CDK and CKIs from whole atria, though contribution of non-myocytes could not be ruled out. These findings indicate that reduction of cyclin and CDK levels, and induction of p27KIP 1 level, are associated with the withdrawal of atrial cell cycle and block the ability of adult cardiomyocytes to re-enter the cell cycle after injury.
The mammalian cell cycle consists of G1, S, G2, and M phases. D-type cyclins are synthesized in early G1 phase, and they bind to and activate CDK4 as cells leave the quiescent phase5ŌĆō7). The cyclin D/cyclin D complex phosphorylates retinoblastoma protein (Rb) in the late G1 phase of cell cycle, canceling Rbs growth-suppressive function and, thereby, facilitating S-phase entry. DNA synthesis occurs in the S phase of cell cycle with activation of cyclin E, A8, 9). Binding of cyclin A or E is necessary for activation CDK2. Cyclin B binds to cyclin B and controls entry into mitosis8, 9). Thus, the previous16) and the present studies indicate that cyclins and CDKs are actively involved in cell cycle progression of cardiomyocytes during the fetal period. There is a rapid transition from G1/M phase of cell cycle to G0/G1 phase of cell cycle in cardiomyocytes during the late gestational and early neonatal periods in experimental animals18, 19).
Yoshizumi el al.,12) and our previous data16), suggested that disappearance of cyclin A was correlated with withdrawal of cardiomyocytes from the cell cycle in human and rat hearts. In addition, the present data indicate that disappearance of cyclin E or B also may be a candidate cyclin for withdrawal of cardiomyocytes from the cell cycle in human atrium. D-type cyclins are synthesized as long as growth factor stimulation is persistent5ŌĆō7). However, they are rapidly degraded when growth factors or mitogens are withdrawn, regardless of the position of the cell in the cell cycle. Growth factors act on adult hearts in an autocrine or paracrine manner20ŌĆō22) and may induce relatively constant D-type cyclins. In this study, adult atria expressed cyclin D1 and D3 proteins. A similar pattern was also observed in rat hearts during development16).
In order to function in the cell cycle, each cyclin must bind to a specific CDK8, 9). Most CDK protein levels and activities were high in atria during the fetal period and those were very low or undetectable levels during the adult periods. Hence, most CDKs could influence the cardiomyocyte cell cycle. PCNA, a DNA polymerase-associated protein, was used as a marker for cell proliferation23). The temporal pattern of PCNA during cardiac development was similar to the pattern of CDKs and was consistent with another report24). However, no significant changes occurred in levels and activities in CDK4 during development in human atrium. It is not clear why there is a species difference between rats and humans16).
The present data indicate that the protein levels of p27KIP 1 increased significantly in the adult atria. The actual nuclear amount of p27KIP 1 in cardiomyocytes may be increased more if we account the decreased ratio of nuclear/cytoplasmic volume between the fetal and adult period25ŌĆō27). Because the amount of p27KIP 1 protein paralleled temporally the inhibition of CDK activity in all instances investigated, the protein abundance is likely the primary mode of p27KIP 1 action8, 9). Therefore, these present data suggest that the high levels of p27KIP 1 protein in atria at the adult period may be a principal inhibitor for cardiomyocyte cell cycle progression after injury. The transgenic animal model, with its selective gene disruption, provides an invaluable tool for understanding the in-vivo function of a selected gene. While all organs have detectable amounts of p27KIP 1, it is most abundant in the thymus and spleen in mice28ŌĆō30) and rats [our laboratory unpublished observation]. Hearts have approximately 20ŌĆō25% of p27KIP 1 protein compared to the spleen in rats [our laboratory unpublished observation]. Transgenic mice lacking p27KIP 1 have grossly normal development but display several phenotypes linked to increased cell proliferation28ŌĆō30) such as increased body size and multiple organ hyperplasia. A positive correlation between the expression amount of p27KIP 1 protein in an organ and the increase in weight of the organ following p27KIP 1 gene disruption was observed. Thus, while there is a 1.8-fold increase of weight in spleen and thymus, 1.2ŌĆō1.3-fold increase of weight in brain and heart were observed in the p27KIP 1-lacking mice29). The increased organ weights result from more increased cell cycle. These results from mice lacking p27KIP 1 strongly support our data that mainly p27KIP 1 limits cell proliferation by inhibiting G1 and S phase cyclin-CDK complexes in cardiomyocytes during development and after injury.
SUMMARY
We have shown that cyclins, CDKs and the CIP/KIP family of CKIs are highly detectable in human atriums during the fetal period and that, differential change occurred at the adult period. Marked reduction of protein levels and activities of cyclins and CDKs are responsible for withdrawal of atrial cardiomyocytes from the cell cycle. Notably, marked induction of p27KIP 1 in adult atrium may be an important inhibitor for blocking the ability of adult cardiomyocytes to re-enter the cell cycle after injury.


 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print





