


INTRODUCTION
Gastric cancer (GC) is a fatal disease that poses a significant health threat. Despite its decreasing incidence rate, it remains a significant global health concern and ranks as the fourth leading cause of cancer-related deaths worldwide [1]. Among the various risk factors associated with GC development, Helicobacter pylori infection is generally accepted as the most critical [2]. H. pylori infection plays a significant role in the perpetuation of chronic inflammation and irritation of the stomach lining, a condition known as gastritis, which leads to the development of premalignant gastric lesions [3].
Recently, there has been growing evidence that gastric bacteria other than H. pylori influence certain aspects of the development of GC [4,5]. In patients with GC, the composition of the gastric microbiota tends to shift in a direction similar to that of the intestinal or oral microbiota [6]. The oral cavity is a crucial route for the continuous supply of bacteria to the upper gastrointestinal tract, including the stomach. Non-H. pylori bacteria originating from the oral cavity may exert a more meaningful effect, especially in gastric environments with low acidity, such as severe mucosal atrophy. Nevertheless, although the gastric microbiome is suspected to play a role in GC development, few studies have focused on its association with the oral microbiome.
Furthermore, most previous studies have solely conducted structural analyses without considering functional aspects, which cannot provide significant insights into the mechanism of gastric carcinogenesis. To elucidate the role of bacteria in the occurrence of GC, concurrent metagenomic functional analyses should yield more revealing results [7].
Therefore, we aimed to characterize the changes in the microbiome during gastric carcinogenesis and assess its predictable functional profiles. Additionally, we aimed to investigate the correlation between the microbiomes of the oral cavity and stomach at different stages of gastric carcinogenesis.
METHODS
Study participants and sample collection
The participants were recruited at Chung-Ang University Hospital between 2017 and 2022. The inclusion criteria for the neoplasm group were patients newly diagnosed with histologically confirmed GC or adenoma, whereas the control group consisted of patients with gastritis only without evidence of GC or adenoma on endoscopic examination. Patients with a history of malignancy, those who underwent prior gastric surgery, those associated with a recent use of antibiotics or probiotics within the last 3 months, or those under 20 years of age were excluded. The final analysis group of 141 participants comprised 132 patients and nine controls. This case-control study was approved by the Institutional Review Board of the Chung-Ang University Hospital (IRB No. C2016047[1790]) and was conducted in accordance with the Declaration of Helsinki. Informed consent was obtained from all participants.
The Vienna Classification System was used to diagnose and categorize gastric neoplasms [8]. Category 3 (non-invasive low-grade adenoma/dysplasia) was classified as the low-grade dysplasia (LGD) group. Category 4.1 (high-grade adenoma/dysplasia) and category 4.2 (non-invasive carcinoma [carcinoma in situ]) were classified as the high-grade dysplasia (HGD) group. Category 4.3 (suspicion of invasive carcinoma) and 5 (invasive neoplasia) were classified as the GC group.
A minimum of 10 mL of saliva was collected by allowing it to accumulate on the floor of the mouth, followed by spitting it into a specimen tube. Samples were stored at ŌłÆ80┬░C until use. Gastric juice samples were obtained according to the following protocol: Patients fasted for more than 8 h before sample collection. For patients who underwent endoscopic examination or resection, a trap tube was connected between the endoscope and the suction tube before the procedure. Gastric juice (7ŌĆō30 mL) was collected by suction through the endoscope at the beginning of the procedure. Gastric tissues were collected from the same gastric sites in both the patient and control groups, following the same protocol. Two samples were obtained from the antrum and two from the body, resulting in a total of four tissue samples, which were then stored at ŌłÆ80┬░C. The endoscopes were washed and disinfected to avoid contamination by immersion in a detergent solution containing 7% proteolytic enzymes and 2% glutaraldehyde before use. Following the collection of gastric juice, the samples were kept at ŌłÆ20┬░C and immediately transferred to the nearby laboratory, without the use of preservative reagents. The collected gastric juice was diluted to 40 mM by the addition of 1 M Tris base and centrifuged at 600 ├Ś g for 10 minutes at 4┬░C to collect the supernatant. After transfer to a new 15 mL tube, it was again centrifuged at 1,500 ├Ś g for 10 minutes at 4┬░C. The supernatant was subsequently harvested and stored at ŌłÆ80┬░C.
DNA extraction and sequencing
All samples were diluted in 10 mL of PBS and incubated for 24 hours prior to DNA extraction. After the centrifugation step at 10,000 ├Ś g for 10 minutes at 4┬░C, the pellet containing the bacteria was diluted with 200 ╬╝L of PBS to create a suspension. Microbial genomic DNA was extracted using a DNeasy PowerSoil kit (QIAGEN, Hilden, Germany) according to the standard protocol provided in the manufacturerŌĆÖs instructions. The isolated genomic DNA was amplified by targeting the V3ŌĆōV4 hypervariable regions of the 16S rRNA gene. This amplification was carried out using specific primers (16S_V3_F: 5ŌĆ▓-TCGTCGGCAGCGTCAGATGTGTATAA GAGACAGCCTACGGGNGGCWGCAG- 3ŌĆ▓ and 16S_V4_ R: 5ŌĆ▓-GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGGACTACHVGGGTATCTAATCC- 3ŌĆ▓). Subsequently, amplicon libraries were prepared from the amplified DNA fragments. The MiSeq Reagent Kit v3 (600-cycle) (Illumina, San Diego, CA, USA) was used to sequence the library reagents. The Nextra XT Index Kit v2 Set A (96 indices, 384 samples) (Illumina) was used for barcodes and adapters. Library preparation for sequencing followed the 16S Metagenomic Sequencing Library Preparation (Part # 15,044,223 Rev. B). All amplicons were sequenced using MiSeq (Illumina) according to the manufacturerŌĆÖs instructions. The bacterial DNA in each sample was quantified using a QIAxpert system (QIAGEN).
Taxonomic assignment and profiling
Paired-end 16S rRNA gene sequences were input into the Quantitative Insights into Microbial Ecology software (QIIME2 v2021. 4) [9]. Adapter sequences were removed using cutadapt. Reads were filtered for quality and chimeric reads using dada2 with manual parameters (trim-left-f 0, trim-left-r 0, trunc-len-f 260, trunc-len-r 200, trunc-q 2, max-ee-f 3, max-ee-r 3) [10]. Taxonomic classification was performed using a Na├»ve Bayes classifier trained on the extracted V3ŌĆōV4 region from the SILVA 138 database. All sequences classified as either chloroplasts or mitochondria were removed.
Statistical analysis
For alpha diversity analysis, samples were rarefied to the minimum read number (1,969) to normalize the read counts. Alpha diversity was further examined by adjusting for age and sex using analysis of variance (ANOVA). Principal coordinate analysis (PCoA) was conducted to determine the individual taxa-level clustering of groups based on the BrayŌĆōCurtis dissimilarity distance and weighted UniFrac distance. The p value for the PCoA was calculated by permutational multivariate analysis of variance (PERMANOVA) using dissimilarity matrices. Additionally, we verified the PCoA after adjusting for age and sex using PERMANOVA. To analyze the differences in microbiome composition and functional profiles between groups, the Wilcoxon rank-sum test was performed. Linear discriminant analysis (LDA) Effect Size (LEfSe) was also used to determine the differentially abundant taxa between the groups for the selection of biomarkers with statistical and biological significance. When converting the feature table to LEfSe format, 100,000 was used as the normalization value. The LEfSe algorithm utilized the KruskalŌĆōWallis test with a cutoff LDA score (log10) of 3.5 for bacteria and 0.25 for functional profiles. During the LEfSe analysis, the false discovery rate (FDR) was applied to further validate the p values. This value represents the q-value and was examined to determine the significance of the results and assess false positives. Additionally, an analysis was conducted using thse multivariate association with linear models (MaAsLin2) package in R, adjusting for age and sex as covariates. A q-value of less than 0.25, adjusted for the FDR using the BenjaminiŌĆōHochberg method, was considered statistically significant. This threshold is the default significance threshold in MaAsLin2, and is commonly used in microbiome studies [11]. To reduce noise, taxa present in less than 10% of all samples with a mean relative abundance of less than 0.01% were removed before conducting LEfSe and MaAsLin2 analyses. For the analysis of the relative abundance and functional profile prediction, the Total-Sum Scaling method was used for normalization. SpearmanŌĆÖs rank correlation coefficient was used to analyze the correlation between the microbiota compositions of all samples. SpearmanŌĆÖs rho and p values were calculated using the cor.test in R [12]. The analyzed correlation coefficient value (rs) was found to be statistically significant when the p value was < 0.05, indicating a meaningful relationship. We performed multiple testing corrections for the Wilcoxon rank-sum test using the BenjaminiŌĆōHochberg procedure to control for the FDR. The functional profiles of the gastric and oral microbiomes of the control and each disease stage were predicted using Tax4Fun2 [7]. All analyses were conducted using R Statistical Software (version 3.6.1; R Foundation for Statistical Computing, Vienna, Austria).
RESULTS
Clinical characteristics of the patients
Samples were collected from 141 patients (nine controls, 58 with LGD, 33 with HGD, and 41 with GC). The study participants had a mean age of 64.4 ┬▒ 11.9 years, with a male proportion of 70.9% (100/141). The clinical characteristics of the enrolled patients according to each sample type are summarized in Table 1.
Changes in the diversity of gastric and oral microbiota during gastric carcinogenesis
We analyzed the microbial diversity in the microbiota of the gastric juice among the four groups. In the alpha diversity analysis of the gastric juice, the Simpson indices were significantly lower in the HGD and GC groups than in the LGD and control groups (p < 0.05; Fig. 1A). In contrast, there was no significant difference in the alpha diversity of the microbial composition within the gastric tissue (both antrum and body) and oral samples among the four groups.
The beta diversity was assessed between the different stages of GC in the gastric juice, tissue (antrum, body), and saliva samples (Fig. 1B). A significant difference in microbiota composition was confirmed among the four groups of gastric juice samples, and the clustering distinction of the control group was particularly prominent (p < 0.001; Fig. 1B). Regarding the beta diversity, there was also a significant difference in the microbial composition of the gastric tissue among the four groups (antrum, p < 0.05; body, p < 0.001; Fig. 1B). Moreover, no significant differences were found in the beta diversity of the oral microbiota among the four groups (p = 0.463, Fig. 1B).
After adjusting for age and sex, the alpha and beta diversity analyses exhibited numerical fluctuations in certain p-values; however, the overall significance remained consistent both before and after the adjustment. Additionally, to clearly demonstrate the differences in beta diversity based on location corresponding to each stage of gastric carcinogenesis, schematic diagrams were constructed (Fig. 1C). Significant differences were observed in microbiome composition among the different sample types at all stages of gastric carcinogenesis. In particular, the difference in microbial composition between the saliva and gastric juice was more pronounced in the control group than in the disease groups (LGD, HGD, and GC groups).
Changes in the gastric and oral microbiota composition during gastric carcinogenesis
A difference was observed in the gastric juice microbial community between the control and disease groups (Fig. 2A). The abundance of Pseudomonadota (formerly Proteobacteria) decreased, while that of Bacillota (formerly Firmicutes) and Actinomycetota (formerly Actinobacteria) increased in the gastric juice of the disease group compared to that of the control group. Moreover, a difference in the microbial composition of the tissues between the control and disease groups (both the antrum and the body) was observed. In the tissues, the abundance of Campylobacterota increased in the disease groups compared to that in the control group. In contrast, no significant difference in the oral microbial composition between the control and disease groups was observed. In the saliva, Bacillota, Actinomycetota, and Bacteroidota predominated in both the control and disease groups, and the composition fractions of each group were very similar, with few differences between the groups.
The LEfSe results for gastric juice samples confirmed significant differences in the abundance of specific strains, including Ralstonia insidiosa, Schaalia odontolytica, Rothia mucilaginosa, Peptostreptococcus stomatis, Streptococcus salivarius, H. pylori, Streptococcus sanguinis, Gemella haemolysans, Streptococcus australis, Streptococcus cristatus, Haemophilus parainfluenza, and Prophyromonas pasteri, among others, within each group (Fig. 2B, q < 0.05). Among these characteristic bacteria, the abundance of R. insidiosa decreased in the disease groups compared to the control group (q < 0.01). However, the abundance of other strains increased in the disease groups than in the control group, with the highest proportion observed in the LGD group (q < 0.05). In the gastric fluid sample analysis, adjusting for sex and age resulted in a loss of significance for some bacterial strains. However, the strains S. odontolytica, P. stomatis, and S. cristatus generally maintained their significance even after adjustment for age and sex (Supplementary Table 1, 2). In the tissues (antrum), the abundance of Pseudomonas yamanorum, H. parainfluenza, and S. cristatus differed significantly between each disease group (q < 0.05). Among the three bacterial species, P. yamanorum and H. parainfluenza were more abundant in the HGD group than in the control group, whereas S. cristatus was the most abundant in the LGD group. In the body tissues, the abundance of Prevotella pallens significantly differed in each disease group (q < 0.05). The abundance increased in the disease group compared to the control group, with the highest observed in the LGD group. In the oral samples, the abundance of Prevotella melanionogenica, G. haemolysans, and Rothia aeria showed significant differences among the disease groups (q < 0.05), with G. haemolysans exhibiting a significantly lower proportion in the control group than in the other disease groups (q < 0.05). Moreover, the bacterial strains exhibited significant changes (q < 0.05) following the LEfSe analysis (Supplementary Table 3ŌĆō8).
Increased similarity between the oral and gastric microbiotas during gastric carcinogenesis
The degree of similarity in bacterial composition among the oral, gastric juice, and gastric tissue samples within each individual disease group was assessed using correlation analysis (Fig. 3, Supplementary Table 9). The similarity between the samples increased as the disease progressed. Moreover, the similarity in microbial composition was relatively low among the different sample types in the control group. The similarity in microbial composition between the saliva and gastric juice samples increased from 0.22 in the control group to 0.58, 0.60, and 0.58 in the LGD, HGD, and GC groups, respectively. Similar tendencies were also found between the gastric juice and tissues (antrum and body), as well as between the saliva and tissues (antrum and body), with higher similarity of microbial composition in the GC groups than in the control group (0.42ŌĆō0.51 vs. 0.30ŌĆō0.27 and 0.68ŌĆō0.66 vs. 0.36ŌĆō0.34, respectively). Meanwhile, the microbial similarity between the antrum and body tissues consistently displayed a stable level of similarity regardless of the disease status. Furthermore, there was a tendency for an increase in the similarity between samples with respect to the stages of gastric carcinogenesis. However, some samples showed a slight decrease in similarity as they progressed from the LGD group, which exhibited the highest similarity, to the HGD and GC groups (GJP-TIA, GJP-TIB, and ORWP-TIB).
Functional profile analysis of the microbiome
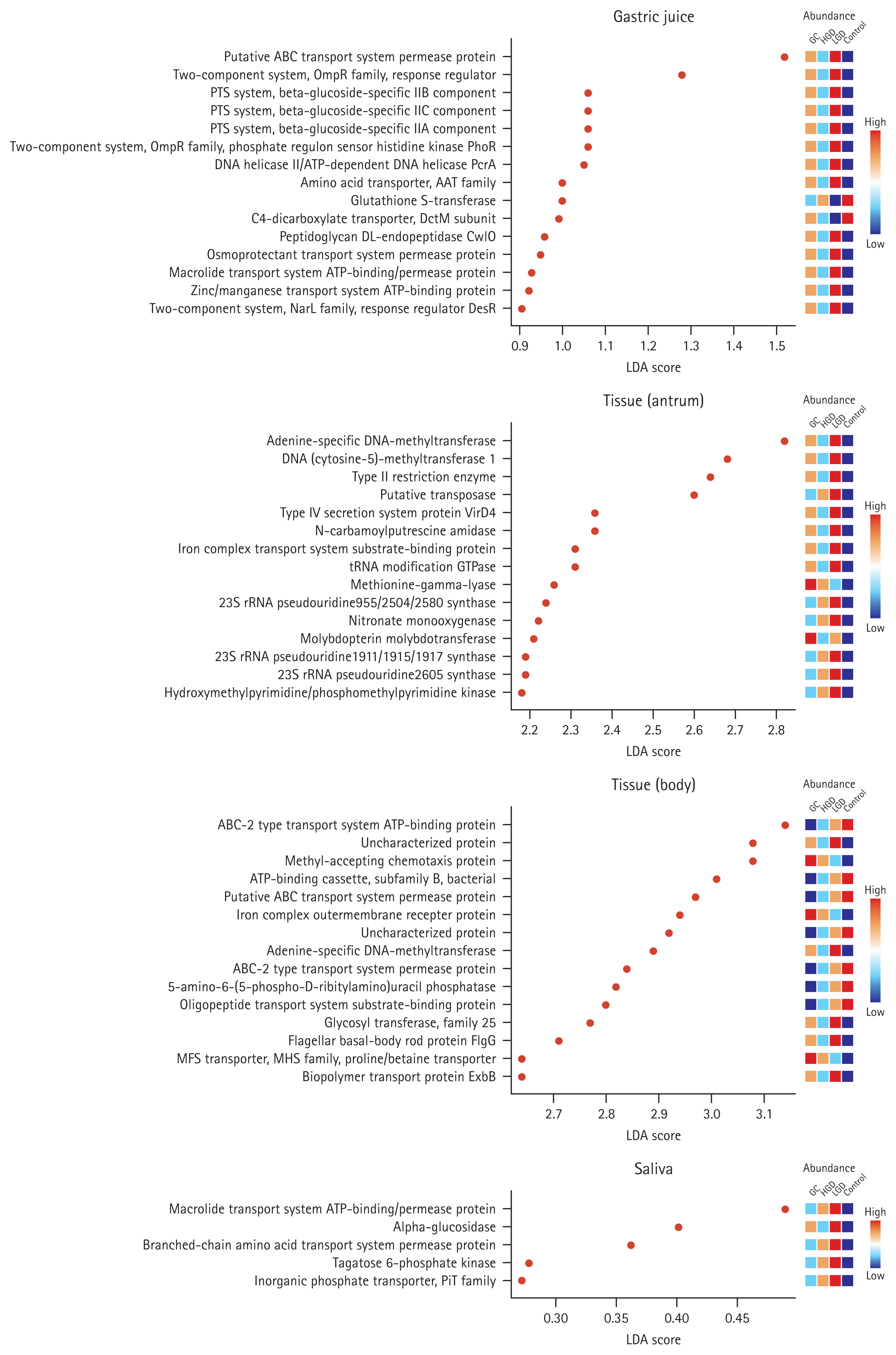
When comparing the functional profiles based on microbiome clustering in four distinct samples across the disease stages, we anticipated that changes would primarily occur in signal transduction (Gastric juice: putative ABC transport system permease protein - K02004, two-component system, OmpR family, response regulator - K02483, PTS system, beta-glucoside-specific IIB component - K02756, PTS system, beta-glucoside-specific IIC component - K02757, PTS system, beta-glucoside-specific IIA component - K02755, two-component system, OmpR family, phosphate regulon sensor histidine kinase PhoR - K07636, two-component system, NarL family, response regulator DesR - K07693, and two-component system, chemotaxis family, chemotaxis protein CheY - K03413; Body tissue: ABC-2 type transport system ATP-binding protein - K01990 and ABC- 2 type transport system permease protein - K01992), DNA replication and expression (Gastric juice: glycine cleavage system transcriptional repressor - K03567; Antrum tissue: adenine-specific DNA-methyltransferase - K07316 and DNA (cytosine-5)-methyltransferase 1 - K00558; Body tissue: adenine-specific DNA-methyltransferase - K07316), and ATP-related pathways (Gastric juice: macrolide transport system ATP-binding/permease protein - K18230 and zinc/ manganese transport system ATP-binding protein - K02074; Body tissue: ATP-binding cassette, subfamily B, bacterial - K06147; Saliva: macrolide transport system ATP-binding/ permease protein - K05685) (Fig. 4, q < 0.05). The functional profiles exhibited significant changes (q < 0.05) in the LEfSe analysis, and this significance was maintained by the application of q-values (Supplementary Table 10ŌĆō13).
DISCUSSION
In this study, we investigated changes in microbial composition and function in the oral cavity and stomach throughout the gastric carcinogenesis pathway. We observed differences in bacterial diversity and composition between the disease and control groups. Furthermore, changes in the metagenomic functional profiles potentially associated with carcinogenesis were observed. We also found that the bacterial composition in the oral cavity and stomach gradually became more similar during gastric carcinogenesis.
Similar to previous studies, the GC group exhibited a decrease in microbial diversity in the gastric juice compared with the control group, with a consistent trend of dominance by specific minority species [5]. The decrease in microbial diversity in various diseases compared to that in the healthy state is well established [13]. Moreover, the beta diversity was significantly different between the healthy and disease groups in the microbial communities of the gastric juice and tissues, indicating variations in the intergroup microbial composition. Specifically, at the phylum level, there was a decrease in the abundance of Pseudomonadota and an increase in that of Bacillota in the gastric juice with disease progression. Moreover, certain species of bacteria showed differences between the control and disease groups. At the phylum level, the abundance of Campylobacterota increased in the disease group compared to the control group. The results observed at the phylum level are consistent with those of previous studies on microbial clustering in GC [14]. Among the specific bacterial species whose abundance increased in the disease group compared to the control group, some have been widely reported to be associated with cancer. S. odontolytica has been identified as a potential pathogen capable of triggering malignant oral conditions [15]. It has also been reported to be associated with higher cancer prevalence [16]. Similar to S. odontolytica, which is commonly found in the oral cavity, S. cristatus and P. stomatis are associated with oral cancer [17ŌĆō19]. Furthermore, P. stomatis has been identified as a potent potential carcinogenesis biomarker not only for oral cancer but also for colorectal cancer [20,21]. In our study, we observed that the abundance of these strains increased in the stomach during gastric carcinogenesis, raising the suspicion that these strains may play a significant role in GC development.
Inspired by the increasing trend of oral microbial strains in the disease group, we further analyzed the correlation between the oral and gastric microbiomes. We compared and analyzed the characteristics of the microbial composition of samples obtained from different anatomical locations in the upper gastrointestinal tract of the patient and control groups. Disparities in the correlations between microbial compositions based on anatomical location were observed between the disease and control groups. In the control group, the oral microbial composition showed relatively low similarity with that of the gastric juice and tissue samples. In contrast, in the disease group, there was an increase in the similarity between the oral and gastric samples, as well as between the gastric juice and tissues. Considering the absence of significant differences in the oral microbial composition among the groups, we suspect that differences in the gastric environment might have altered the process by which oral bacteria influence the microbial composition upon entering the stomach. The oral cavity serves as a continuous route for bacterial influx into the upper GI tract. The bacterial population in the esophagus originates from the oral cavity, and oral bacteria may influence the development of esophageal cancer. Similarly, it can be inferred that oral bacteria can significantly impact the gastric microbial composition [22ŌĆō25].
Infection with H. pylori, a well-recognized risk factor for GC, leads to persistent chronic inflammation of the gastric mucosa, eventually causing mucosal atrophy and worsening atrophic gastritis. As atrophic gastritis progresses, gastric acid secretion decreases, which can substantially affect the gastric microbial composition [26]. Bacterial strains that would typically struggle to thrive in the highly acidic environment of the stomach could proliferate more easily as the acidity decreases. This could also account for the increased similarity between the floating bacteria in the gastric juice and the clustered bacteria adhering to the gastric mucosa in the stomach of patients with gastric neoplasms. Previous studies have demonstrated this phenomenon, and the altered gastric microbial composition in patients taking proton pump inhibitors further supports this observation. Therefore, although a significant proportion of orally ingested microbial strains may persist in a healthy stomach, these strains may more readily establish clusters and microbial compositions within the stomach as gastric carcinogenesis progresses, coupled with reduced gastric acid secretion. Recent studies have actively explored the potential impacts of non-H. pylori gastric bacteria on GC development, which aligns with this hypothesis.
Consequently, additional analyses were conducted to analyze the functional profile prediction and assess the relevance of the oral-gastric microbiome cluster. The functional profiles of microorganisms known to be associated with cancer, such as the ATP-binding cassette transport system, two-component system, and phosphoenolpyruvate-carbohydrate phosphotransferase system, showed significant changes in the LEfSe analysis when compared between the disease and control groups. These profiles were higher in the disease group than in the control group, suggesting a positive correlation, similar to the observed increase in P. stomatis and S. cristatus with disease progression. This indicates that increased microbial metabolism may facilitate the survival and growth of these organisms. The aforementioned functional profiles, such as the ABC transport system and two-component system, operate in bacteria. The ABC transport system can also function in eukaryotic cells, aiding nutrient uptake, toxin secretion, and sometimes chemotherapy drug resistance in tumor cells [27ŌĆō29]. The two-component system serves as a central player in bacterial signal transduction, catalyzing DNA binding, and biochemical reactions. It is involved in various physiological functions related to survival, such as sporulation and antibiotic resistance, making it play a role in inducing cancer [30,31].
Our study had some limitations. In some sample groups, there was an imbalance in the distribution of sex and age, with a bias towards one side. Consequently, we included groups with sex and age imbalances in the analysis. After conducting the analysis, additional verification was performed after adjusting for age and sex. Although there were no significant changes in alpha and beta diversity, some bacterial strains lost significance in the LEfSe results when adjusted for sex and age. However, in gastric juice samples, the significance of S. odontolytica, P. stomatis, and S. cristatus strains was maintained. This may have influenced the results of the metagenomic analyses between the control and disease groups. In the analysis of functional profiles, we conducted a predictive analysis rather than an actual functional analysis. The current body of evidence often lacks comprehensive analyses of both structural and functional aspects of microbial communities. Therefore, it is not possible to conclusively demonstrate the precise role and mechanism of microbial communities in GC development. Despite these limitations, our study revealed some interesting findings.
Our findings suggest that compositional changes occur in the gastric microbiome, accompanied by functional implications during carcinogenesis. This indicates that microbes other than H. pylori, a prominent contributor to GC, play a role in altering the balance between the host and commensal microbial flora within the stomach. The increased similarity between the microbiota of the oral cavity and stomach implies that the oral microbiome might play a role in carcinogenesis, sustaining the inflammatory process and heightening the risk of GC development through its distinctive genotoxic potential.
KEY MESSAGE
1. The correlation between oral and gastric microbiota increases during gastric carcinogenesis, implying a possible link between oral bacteria and GC.
2. The abundance of certain specific oral commensals, known to be associated with cancer development, was increased in the gastric juice of patients with LGD and GC compared to controls.
3. Functional profile analysis of the microbial composition of the gastric juice further supported the potential role of the microbiome in gastric carcinogenesis.




 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Supplement table 1
Supplement table 1 Print
Print





