


Luteinizing hormone (LH) is a member of gonadotropin secreted by adenohypophysis, which plays an important physiological role in male pubertal development and reproductive function [1]. Idiopathic hypogonadotropic hypogonadism (IHH) in men due to selective LH deficiency is extremely rare. Weiss et al. [2] described for the first time in 1992 that the mutation of the luteinizing hormone β-subunit (LHB) gene can cause selective LH deficiency. At present, 10 male patients with LHB mutations have been reported globally, most of them showed delayed pubertal development and abnormal spermatogenesis [2–9]. In this study, the next-generation sequencing (NGS) combined with Sanger sequencing technology was used for the first time to screen and verify a new mutation site in the LHB gene.
The patient reported in this study was a 35-year-old Chinese man, who sought treatment for primary infertility and azoospermia that had been ongoing for 3 years after marriage. He reported decreased sexuality but was able to complete intercourse. Although his volume was normal but no sperm was found. He was 170 cm tall and weighed 90 kg, physical examination revealed sparse beard and facial hair, normal distribution of armpit and pubic hair, penis length 7 cm, testis volume 2 mL, no palpable abnormalities were found in bilateral spermatic cord and vas deferens. The results of initial and subsequent laboratory tests were as follows: undetectable LH level (< 0.100 mIU/mL; reference range, 1.7 to 8.6), a low serum testosterone (T) level (1.51 ng/mL; reference range, 2.49 to 8.36), and an elevated follicle stimulating hormone (FSH) level (16.94 mIU/mL; reference range, 1.5 to 12.4). The inhibin B level was less than 10 pg/mL (reference range, 18.22 to 311.270). The levels of prolactin, estradiol, thyrotropin, adrenocorticotropic hormone, and anti-Mullerian hormone were normal.
After giving 100 μg gonadotropin-releasing hormone for 90 minutes, the FSH level increased to 25.47 mIU/mL, whereas LH remained undetectable. The serum T level increased from 1.51 to 3.29 ng/mL after human chorionic gonadotropin (hCG) 5,000 IU injection. Magnetic resonance imaging of the brain and pituitary showed no abnormalities. His karyotype was 46 XY, and no site deletion was found in Y chromosome microdeletion. The initial diagnosis was IHH caused by selective LH deficiency. hCG 2,000 IU was injected subcutaneously three times a week. After 3 months of treatment, his weight increased from 90 to 105 kg, and he developed sleep snoring and often felt suffocated after waking up. Then, he was transferred to a specialist otolaryngologist and diagnosed with sleep apnea syndrome. Therefore, the patient refused to use any hormone therapy.
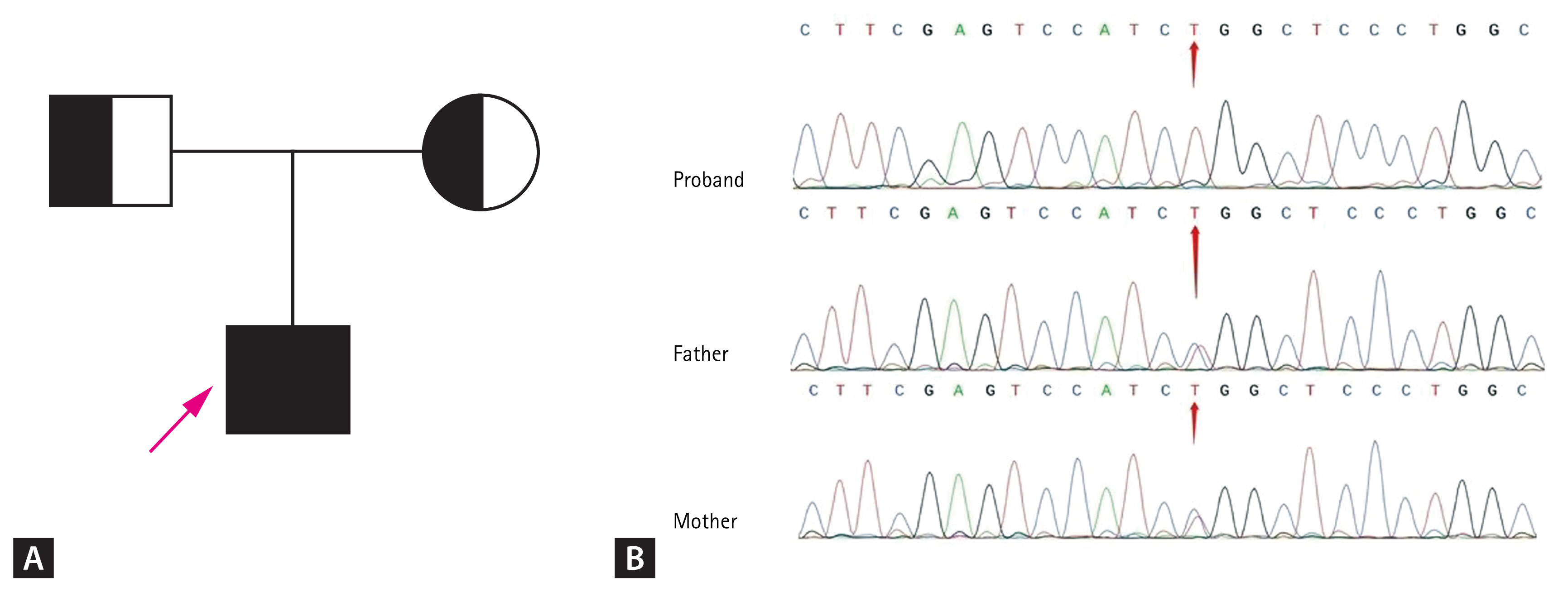
The blood of the patient and his parents was collected after obtaining informed consent from the patient. Then, the patient’s peripheral blood DNA was extracted for whole-exome sequencing, and the obtained data were analyzed bioinformatically. A new missense mutation was found at exon 2: c.262 C>T (p.R88W) of the LHB gene, which was also confirmed by Sanger sequencing. The patient’s parents were heterozygous carriers of this mutation (Fig. 1).
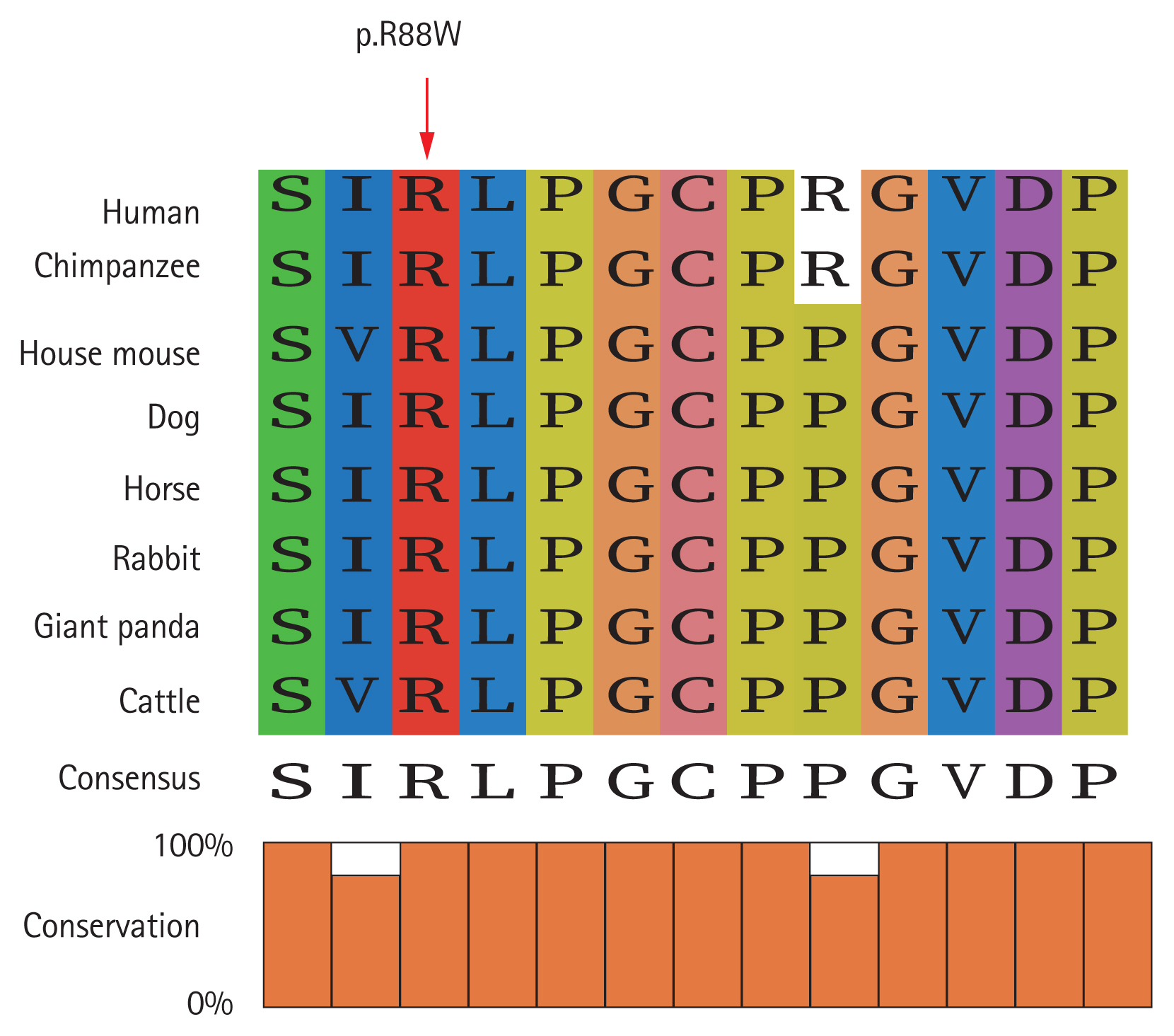
Computational analysis by the online variant of pathogenicity prediction tools showed that the LHB sequence mutation was most probably a disease-causing mutation (Table 1). In the ExAC and gnomAD databases, the allele frequency of the c.262C>T: p.R88W variant was very low (Table 1), indicating the rarity and pathogenicity of this mutation. The variant was highly conserved from human to cattle as revealed by sequence alignment analysis (Fig. 2).
LH is a heterodimer composed of a common alpha subunit and a hormone-specific beta subunit of the glycoprotein hormone family, in which the LHB is expressed by the LHB gene [1]. The mutations reported so far are all located in the β subunit and are more common in men. At present, eight different LHB gene mutations have been described in male selective LH deficiency, half of which occurred in consanguineous families. These genotypic abnormalities included the following: IVS2 + 1G→C mutation, which induced a gross abnormality in the processing of LHB mRNA [3]; deletion of nine bases in exon 2, which weakened the cystine knot folding motif [4]; c118_120del, which resulted in the loss of lysine at position 40 of the mature peptide [5]; and c.84G>A nonsense mutation, producing a truncated LHB peptide [6]. The genotypes of other sporadic cases were as follows: a missense mutation, which eliminated the receptor-binding activity [2]; a homozygous missense mutation in exon 2, which prevented the binding of heterodimer hormone to the receptor [7]; a compound heterozygous mutation, which impaired the maturation of LHB peptide and disrupted splicing of the mRNA [8]; and a point deletion of the thymine nucleotide at position 215 in exon 3, which would cause a frameshift and premature termination at codon 128 [9].
In this study, a sporadic case was reported with clinical manifestations of azoospermia and abnormal gonadal development (mainly sparse beards and small testicles). Laboratory tests showed that LH could not be detected, the serum T level decreased, and the inhibin B level was very low. The NGS was used to screen for gene mutations. The 262 base C of the coding region of the LHB gene was found to be mutated T, resulting in the substitution of arginine to tryptophan at the 88th amino acid position of the encoded protein, which might inactivate LH biologically by weakening the binding of heterodimer hormone to its receptor. For the treatment of young men with LH deficiency due to LHB mutations, it is currently believed that gonadotropins rather than testosterone should be initially used to promote Sertoli and Leydig maturation and improve spermatogenesis [10]. Therefore, treatment for the patient with hCG was initiated. Unfortunately, the patient stopped the treatment midway because of sleep apnea caused by abnormal weight gain.
In conclusion, we first tried to use NGS to screen for selective LH deficiency gene mutations, and validated a new homozygous missense mutation in LHB gene. The limitation of this study was that the functional analysis of the mutant β-subunit was not performed. This study also confirmed that NGS technology could be used to screen the genetic causes of rare diseases.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print





