


INTRODUCTION
Heart failure (HF) affects approximately 26 million people worldwide, with more than one million patients hospitalized every year in the United States and Europe [1]. In the United States, HF accounted for one out of nine deaths in 2013 [2]. Owing to the aging population, the prevalence of HF is increasing [3]. Despite the development of new pharmacotherapy agents for HF, it remains a serious health problem and is associated with high mortality and significant medical costs [4,5].
The goals of therapy for patients with advanced HF are to improve survival, slow disease progression, and alleviate symptoms. Renin-angiotensin-aldosterone system (RAAS) blockers, such as angiotensin converting enzyme (ACE) inhibitors and angiotensin receptor blockers (ARBs), are commonly used to achieve these goals; they decrease preload and make it easier for the heart to pump blood. However, the use of ACE inhibitors is limited by the common side effects, such as cough; therefore, ARBs are potential alternative treatments [6].
Fimasartan is the ninth and the most recent ARB to be approved by the Korean Food and Drug Association as an antihypertensive agent [7-9]. In previous studies, fimasartan was shown to exert protective effects in animal models of myocardial ischemia: the suppression of apoptosis [10] and the prevention of intimal thickening and plaque rupture [11]. Furthermore, fimasartan has been reported to prevent doxorubicin-induced cardiomyopathy and improve survival [12]. Therefore, fimasartan is regarded as a potential candidate for the attenuation of HF.
Recently, in vivo chemical screening in a zebrafish model of HF has emerged as a rapid and efficient method to identify lead compounds that modulate specific biological processes [13,14]. We have also previously reported a zebrafish model of dilated cardiomyopathy induced by brief treatment with terfenadine [15].
In this study, we hypothesized that treatment with fimasartan exerted protective effects against the development of HF and improved survival through the prevention of apoptotic cell death in a terfenadine-induced in vivo zebrafish model of HF.
METHODS
Zebrafish husbandry and breeding
Zebrafish (Danio rerio) embryos were maintained in egg water at 28.5┬║C in an automatic circulating tank system in accordance with instructions from The Zebrafish Book [16]. Adult fish were maintained at 28┬░C under a 14:10 hours light:dark cycle, and were fed three times daily with Artemia (INVE, Dendermonde, Belgium). To visualize the heart chambers, a transgenic strain of zebrafish larvae that expresses green fluorescent protein (GFP) exclusively in the cardiac myosin light chain 2 (cmlc2) [Tg (cmlc2:gfp)] was used [17]. Tg (cmlc2:gfp) lines were kindly provided by the zebrafish Organogenesis Mutant Bank in Korea. All animal study protocols were approved by the Institutional Animal Care and Use Committee of Seoul National University (SNU-150330-3).
Chemical treatment of zebrafish
To maintain the optical clarity of embryos, the egg water was supplemented with 0.003% 1-phenyl-2-thiourea (Sigma, St. Louis, MO, USA) at 24 hours post-fertilization (dpf). For all experiments excluding survival analysis, the larvae were transferred into 6-well microplates and egg water was replaced with a solution containing 20 ╬╝M terfenadine (Sigma) or vehicle (0.1% dimethyl sulfoxide [DMSO]) for 12 hours at 3 days dpf. After terfenadine treatment, fimasartan (Boryung Pharm Co. Ltd., Seoul, Korea) was added to the treatment group for 12 hours before the analysis was performed.
Cardiac morphology
After 12 hours treatment with fimasartan, wild-type zebrafish were anesthetized with 0.04% tricaine (Sigma) for 1 minute and images were obtained by using a stereomicroscope (M165 FC, Leica, Wetzlar, Germany) for morphologic observation. Tg (cmlc2:gfp) larvae were immersed in 0.15% low-melt agarose and transferred to a confocal dish to visualize the movement of cardiac chambers by using a fluorescence microscope (Leica AF2000). The atrial area was calculated by using ImageJ graphical analysis software (National Institutes of Health, Bethesda, MD, USA). At minimum, five different larvae in each group were analyzed.
Functional assessment
Fractional shortening (FS) was calculated from the ventricular diastolic and systolic diameters, as measured from time-lapse images collected intervals of 0.16 seconds. Long-axis diameters were acquired from still images by using ImageJ software and FS was calculated from the following formula: FS = (VDD ŌĆō VSD) / VDD ├Ś 100% (VDD, ventricle diastolic diameter; VSD, ventricle systolic diameter).
The blood flow and time interval between heartbeats were also assessed. After exposure to the drug, the larvae were anesthetized with 0.04% tricaine for 1 minutes, transferred to a confocal dish, and visualization by using a stereomicroscope (Leica M165 FC) fitted with a highspeed video camera. The orientation of the larva was then adjusted very gently so that the larva lay on its side with its head to the left. First, the camera was positioned to capture the whole heart at a rate of 30 frames per seconds (fps), and then repositioned to capture the dorsal aorta and caudal to the swim bladder at 120 fps. The camera in both positions was independently focused on the respective regions of interest to ensure optimal image quality, and set to record simultaneously.
To determine atrioventricular (AV) synchrony, videos of the heart were analyzed by using MicroZebraLab version 3.5 (ViewPoint, Lyon, France). The arrhythmic rate was calculated by using the following formula: arrhythmic rate (%) = VBR/ABR (VBR, ventricular beat rate; ABR, atrial beat rate). The blood flow videos were also analyzed by using ZebraBlood v1.3.2 (ViewPoint), which detects changes in heartbeat intervals.
RNA isolation and quantitative real-time PCR
Total mRNA was extracted from 20 larvae per treatment group by using TRIzol reagent (Invitrogen, Carlsbad, CA, USA) in accordance with the manufacturerŌĆÖs instructions. One microgram of isolated RNA was reverse-transcribed into cDNA by using M-MLV reverse transcriptase (Enzynomics, Daejeon, Korea). Quantitative PCR was performed by using SYBR Green qPCR Master Mix (Applied Biosystems, Foster City, CA, USA) and an ABI real-time PCR 7500 machine (Applied Biosystems). All samples were normalized to the mRNA expression of ╬▓-actin. The primers used were as follows: natriuretic peptide b (Nppb) forward primer: 5ŌĆÖ-CAT GGG TGT TTT AAA GTT TCT CC-3ŌĆÖ; Nppb reverse primer: 5ŌĆÖ-CTT CAA TAT TTG CCG CCT TTA C-3ŌĆÖ; p53 forward primer: 5ŌĆÖ-GGG CAA TCA GCG AGC AAA-3ŌĆÖ; p53 reverse primer: 5ŌĆÖ-ACT GAC CTT CCT GAG TCT CCA-3ŌĆÖ; ╬▓-actin forward primer: 5ŌĆÖ-TGG TGA CCT GAC AGA CTA CCT GAT-3ŌĆÖ; ╬▓-actin reverse primer: 5ŌĆÖ-CGG ACA ATT TCT CTT TCG GCT GTG-3ŌĆÖ.
TUNEL assay
The fragmentation of DNA in apoptotic cells was identified by the terminal deoxynucleotide transferase-mediated dUTP nick-end labeling (TUNEL) assay using the In Situ Cell Death Detection Kit, POD (Roche Diagnostics GmbH, Mannheim, Germany). Zebrafish larvae were fixed in 4% paraformaldehyde (PFA) overnight at 4┬░C. After fixation, excess 4% PFA was removed by five rinses in phosphate-buffered saline with Tween 20 (PBST), each of 5 minutes, and 100% methanol was added. The larvae were incubated at ŌĆō20┬░C for at least 30 minutes, and rehydrated in a graded methanol series (75%, 50%, 25%, and twice in 0%). The larvae were then incubated with proteinase K (50 ╬╝g/mL in PBST) for 30 minutes at room temperature (RT), and excess proteinase K was removed by two rinses in PBST, each of 5 minutes. The larvae were re-fixed in 4% PFA for 20 minutes at RT and excess 4% PFA was removed by five rinses with PBST, each of 5 minutes, and then post-fixed by the application of a 2:1 mixture of pre-chilled ethanol and acetic acid for 10 minutes at ŌĆō20┬░C. After three rinses with PBST, each of 5 minutes, the larvae were incubated for 2 hours at 37┬░C in labeling solution, which consisted of terminal deoxynucleotidyl transferase (TdT) and fluorescein-conjugated deoxynucleotide in buffer. After three rinses with PBST, each of 5 minutes, the larvae were incubated for 30 minutes in a converter-peroxidase (POD) in a humidified chamber at 37┬░C. Finally, excess converter-POD was removed by three rinses with PBST, each of 5 minutes, and apoptotic cells were visualized by using metal-enhanced DAB (Dako, Carpinteria, CA, USA).
Survival and motility assay
For the survival assay, larvae at 3 dpf were immersed in 20 ╬╝M terfenadine for 12 hours, and 200 ╬╝M fimasartan was added for 60 hours before analysis. For the motility assay, larvae at 3 dpf were immersed in 20 ╬╝M terfenadine for 24 hours and the medium was replaced with egg water containing 200 ╬╝M fimasartan for 24 hours in the treatment group. The larvae were transferred to 6-well microplates in egg water and allowed to move freely at room temperature. The embryo positions were recorded every 0.5 second for 20 seconds to generate a trace.
Statistical analysis
Continuous variables were shown as the mean ┬▒ standard deviation and assessed by using the StudentŌĆÖs t test and analysis of variance (ANOVA). Categorical variables were presented as numbers and percentages and were assessed by using the chi-square test, with Kaplan-Meier analysis used for the comparison of survival. Statistical analyses were computed by using SPSS version 25 (IBM, Chicago, IL, USA) and figures were drawn by using an open-trial version of GraphPad Prism version 5 (Graph- Pad Software, San Diego, CA, USA). A p value of < 0.05 was deemed statistically significant and is indicated in the figures by an asterisk; p values of < 0.01 and < 0.001 are indicated by two and three asterisks, respectively.
RESULTS
Fimasartan ameliorates terfenadine-induced HF in zebrafish
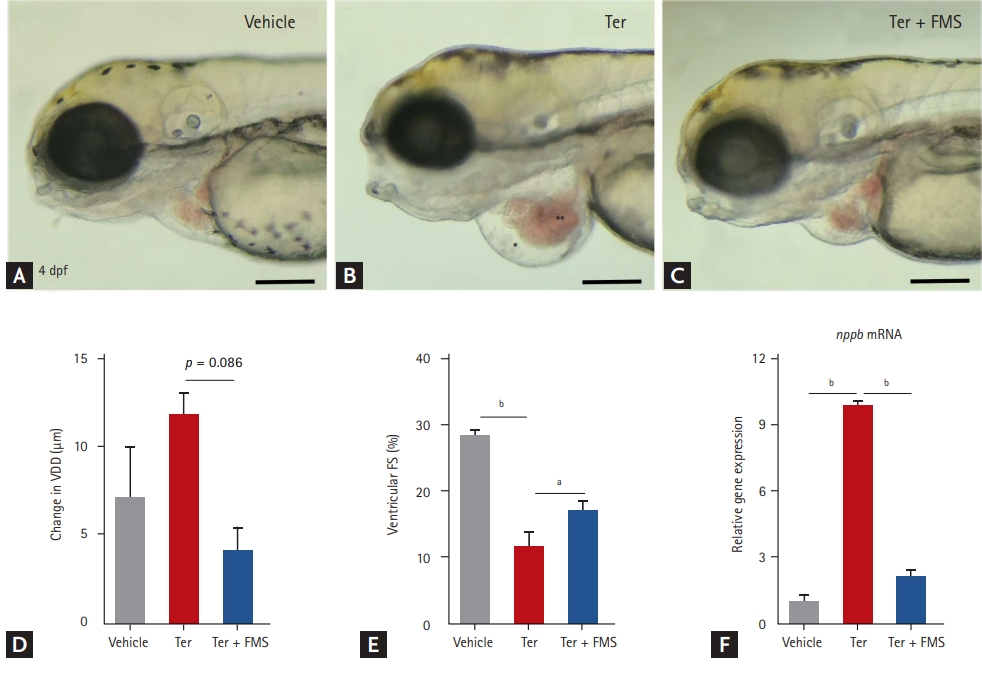
Zebrafish larvae at 3 dpf were treated with either 20 ╬╝M terfenadine or vehicle (0.1% DMSO) for 12 hours. In the fimasartan treatment group, 200 ╬╝M fimasartan was added and the larvae were incubated for 12 hours. Consistent with our previous study [15], zebrafish treated with terfenadine for 24 hours developed progressive HF, with dilatation of the ventricle, venous congestion, and decreased ventricular contractility as measured by FS (Fig. 1A-1E). Nppb mRNA expression was higher in the terfenadine-treated larvae than in vehicle-treated larvae (Fig. 1F). These findings indicated that transient treatment with terfenadine induced HF in zebrafish and reproduced the characteristics of human patients with HF, such as dilatation of the cardiac chambers and decreased ventricular function.
Treatment with fimasartan reduced ventricular dilatation and nppb mRNA expression induced by terfenadine (Fig. 1C and 1F). Ventricular function, as assessed by FS, was also significantly preserved in the fimasartan treatment group compared with the terfenadine only group (16.9% ┬▒ 3.1% vs. 11.4% ┬▒ 5.6%, p < 0.05).
Fimasartan promotes recovery of cardiac output
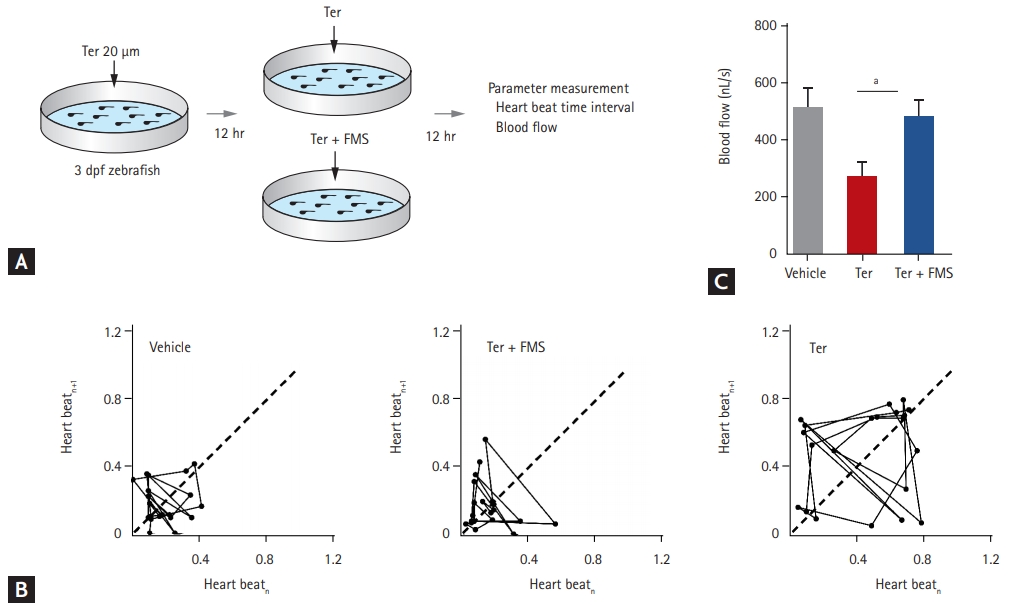
To explore the physiologic effect of fimasartan in the zebrafish model of HF, hemodynamic parameters were measured, such as heartbeat interval and blood flow velocity (Fig. 2A). To assess the variability in heart rate, Poincar├® plots were produced by using two consecutive time intervals between heartbeats. In control larvae treated with vehicle only, the data points in the plot were concentrated between 0.01 and 0.30 second, clustering at approximately 0.19 second. In contrast, terfenadine-treated larvae resulted in a dispersed plot, deviating from the control axis (in the range from 0.10 to 0.80 second). Heartbeat variability significantly increased from 0.19 ┬▒ 0.12 second in vehicle-treated larvae to 0.46 ┬▒ 0.27 seconds in terfenadine-treated larvae (p < 0.001). Treatment with fimasartan markedly reduced the variability, to 0.16 ┬▒ 0.13 second (p < 0.001), with heartbeat intervals in the range from 0.08 to 0.40 second (Fig. 2B).
Blood flow velocity was lower in terfenadine-treated larvae than in vehicle-treated larvae, whereas fimasartan treatment preserved blood flow velocity. In detail, blood flow velocity in the terfenadine-treated larvae decreased from 507.0 ┬▒ 170.6 to 273.0 ┬▒ 109.0 nL/sec, whereas treatment with 200 ╬╝M fimasartan resulted in marked preservation of blood flow velocity, with a value of 479.1 ┬▒ 124.1 nL/sec (Fig. 2C). The response of blood flow velocity to fimasartan treatment was also dose-dependent (Supplementary Fig. 1). These results indicated that fimasartan could reverse the variability in heartbeat and the decrease in blood flow velocity induced by terfenadine.
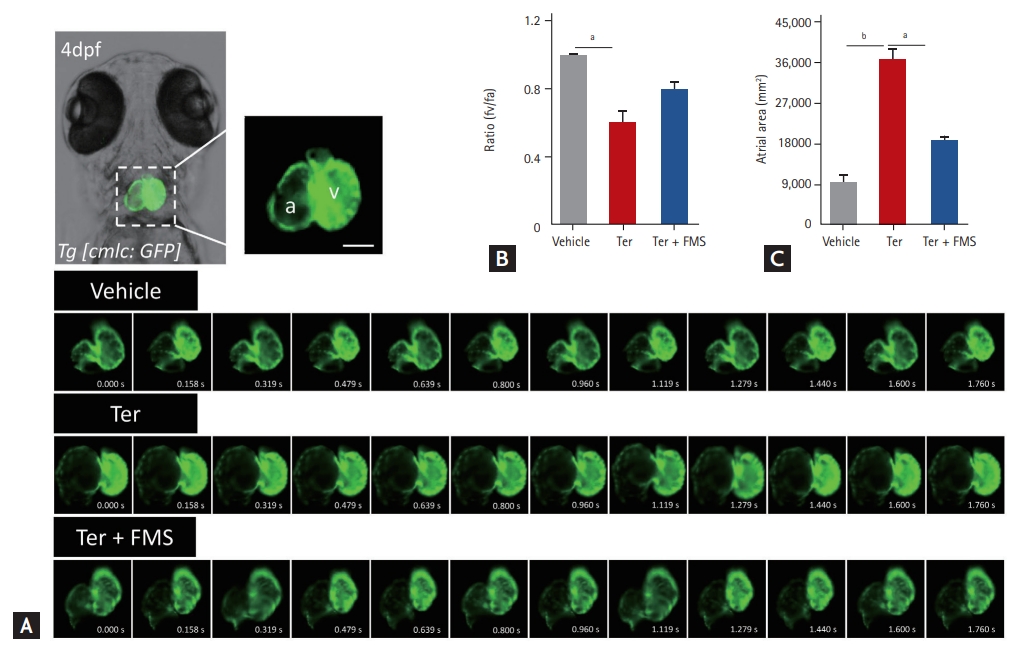
Fimasartan improves atrioventricular dyssynchrony
We previously reported AV dyssynchrony in zebrafish after treatment with terfenadine [15]. We investigated contraction timing, rhythmic rate, and atrial area to confirm whether fimasartan improved terfenadine-induced AV dyssynchrony. The atria and ventricles of Tg (cmlc2:gfp) larvae were clearly visible, and contraction times were discriminated by using a series of images taken by in vivo time-lapse video recordings. In contrast with the synchronous contraction and dilatation of the atria and ventricles observed in vehicle-treated larvae, the larvae treated with terfenadine showed dyssynchronous AV movement (Fig. 3A and Supplementary Video 1). The AV ratio of terfenadine-treated larvae was approximately 0.6, and the atrial area was significantly increased. Unexpectedly, in larvae treated with fimasartan, atrial contraction was more regular and the AV ratio was preserved, with a value of approximately 0.8 (Fig. 3B). The atrial area was also significantly smaller than that in the terfenadine-treated group. The atrial areas of terfenadine-treated larvae were increased four-fold compared with the vehicle-treated larvae, from 9,080.2 ┬▒ 1,646.6 to 36,293.8 ┬▒ 2,322.5 ╬╝m2, whereas the larvae treated with fimasartan only showed a two-fold increase, to 18,500.8 ┬▒ 807.4 ╬╝m2 (Fig. 3C).
Fimasartan prevents apoptotic cell death
To evaluate whether fimasartan reduced apoptotic cell death in terfenadine-induced zebrafish HF, an in situ TUNEL assay was performed to visualize apoptotic cells. Increased chromatin fragmentation, indicating cell death, was observed around the heart field in terfenadine-treated larvae, and this was suppressed by fimasartan treatment (Fig. 4A-4C). The quantification of TUNEL-positive cells also yielded similar results, which showed that fimasartan suppressed chromatin fragmentation induced by terfenadine (Fig. 4D). Transcription of the p53 gene was also significantly increased in terfenadine-treated larvae, whereas fimasartan-treated larvae showed only a mild increase in p53 mRNA expression. These findings indicated that fimasartan significantly suppressed terfenadine-induced apoptosis in an in vivo zebrafish model of HF.
Fimasartan improves zebrafish motility and survival
The motility of zebrafish larvae was dramatically reduced by terfenadine treatment, but showed near-normal values in larvae treated with fimasartan. The quantification of motility by using video analysis showed a significant difference between terfenadine-treated and fimasartan-treated larvae (9.58 ┬▒ 6.89 mm vs. 36.97 ┬▒ 22.79 mm, p < 0.001) (Fig. 5A-C and Supplemental Video 2). Fimasartan treatment also significantly reduced the mortality of terfenadine-treated zebrafish (terfenadine + fimasartan 36.0% vs. terfenadine 96.0%, p < 0.001) (Fig. 5D).
DISCUSSION
In this study, treatment with fimasartan effectively prevented the development of HF and improved survival in our zebrafish model. First, HF was induced by treatment of terfenadine for 12 hours, as denoted by marked dilatation of cardiac chambers and decreased ventricular systolic function; in contrast, treatment with fimasartan reversed changes in heart size and enhanced its function. Second, fimasartan treatment effectively preserved cardiac output, reduced heartbeat variability, accelerated blood flow, and decreased atrial area. Finally, fimasartan treatment also improved mobility and the survival of terfenadine-treated zebrafish. These results suggested that fimasartan may be a candidate pharmacotherapy agent for HF.
In the current study, an in vivo zebrafish model of terfenadine-induced cardiomyopathy was used to examine the protective effects of fimasartan in HF. By using this model, we were able to assess the entire scope of HF pathophysiology. At the cellular level, the mRNA expression of natriuretic peptide was congruent with treatment status: it was increased in terfenadine-treated zebrafish, but was decreased by fimasartan treatment. The physiologic status was assessed by the time interval between heartbeats and the mean blood flow velocity. We were also able to assess functional status through the measurement of FS and motility by using video analysis. Finally, p53 mRNA expression and chromatin fragmentation by TUNEL assay was used to assess cell survival.
Whenever cardiac output falls, compensatory mechanisms are induced in an attempt to maintain blood pressure and organ perfusion. For this reason, RAAS is a well-established therapeutic target in the treatment of HF. However, angiotensin II receptor antagonists are recommended only when ACE inhibitors are intolerable or contraindicated. Previous clinical studies have shown that losartan [18], valsartan [19], and candesartan [20] may improve outcomes and reduce hospitalization in patients HF; however, there have been no studies on the use of fimasartan.
Previous studies have reported that terfenadine induces cardiac toxicity with bradycardia and arrhythmia in zebrafish larvae [12,21,22]. Moreover, recent studies have confirmed that terfenadine-treated zebrafish larvae can be used as a model to screen for positive inotropes [13]. The features of cardiac dysfunction in zebrafish larvae induced by terfenadine treatment are similar to those in human HF. In addition to triggering atrioventricu lar dyssynchrony and venous congestion in zebrafish, terfenadine has also been reported to cause ventricular tachycardia and fibrillation in rabbits [23], which may result in the rapid decline of ventricular function and the development of HF [24]. In the current study, we treated zebrafish larvae at 3 dpf with 20 ╬╝M terfenadine for 12 hours to induce dilated cardiomyopathy. As expected, the larvae showed an increase in cardiac dimensions and a decrease in systolic function. In addition, blood flow velocity was decreased and contraction time was delayed. This decrease in blood flow can result in hypoperfusion, and there have been several reports that renal hypoperfusion activates the RAA system in patients with HF [25-27]. Reduced cardiac output in early HF induces RAAS-activated fluid retention, which subsequently increases ventricular preload and cardiac output as a compensation method. However, if ventricular dysfunction progresses, compensation mechanisms fail and venous pressure increases [28]. As we have demonstrated similar mechanisms in zebrafish larvae, we believe that our terfenadine-induced model of dilated cardiomyopathy can be used to test the effectiveness of RAAS inhibitors.
Fimasartan, an ARB, has been approved for the treatment of essential hypertension in Korea. In addition to its angiotensin II-blocking effects, fimasartan has shown superior inhibition of the contraction of isolated rabbit thoracic aorta compared with other ARBs, such as losartan and candesartan [29], and has been reported to have protective effects in a porcine model of acute myocardial infarction [30]. Fimasartan was also reported to reduce ischemic cell death when used to treat transient focal ischemia in rats [31], and to prevent unilateral ureteral obstruction-induced apoptosis in mice [32]. Although fimasartan has not been previously studied for the treatment of HF, it is expected to exert similar effects to other ARBs, and potentially greater efficacy because of its higher affinity for angiotensin II type 1 (AT1) receptors compared with other drugs of its class in preclinical studies [33]. The AT1 receptor is involved in the classical physiological actions of angiotensin II: the regulation of blood pressure, electrolytes, and water balance; thirst; hormone secretion, and the regulation of renal function [34]. Therefore, the inhibition of angiotensin II signals can dilate arteries and veins, thereby reducing arterial pressure, decreasing preload and afterload, and increasing blood flow velocity and FS.
In line with previous studies that showed the potent attenuation of myocardial apoptotic cell death by fimasartan in reperfused rat hearts and H9C2 cells [10], we showed that treatment with fimasartan prevented apoptotic cell death. The activation of the AT1 receptor enhances the influx of intracellular calcium and stimulates calcium-dependent endogenic endonucleases, which cause DNA laddering, cell shrinkage, and the formation of apoptotic bodies [10]. In addition, AT1 receptor blockers may exert cardioprotective actions in addition to their ability to lower blood pressure, such as anti-apoptotic, anti-atherosclerotic, and target organ-protecting effects [35]. Our study also showed that blocking the AT1 receptor decreased the number of apoptotic cells and pro-apoptotic p53 expression in zebrafish larvae (Fig. 4). Interestingly, p53 induces apoptosis in cardiac myocytes via the activation of classical renin-angiotensin system. This may be limited to myocytes because the induction of p53 is insufficient to trigger apoptosis in other cell types [36].
Randomized trials have shown that ARBs improve survival in patients with HF who are intolerable to ACE inhibitors [37-39]. However, there have been reports that elderly patients with HF treated with losartan had poorer survival rates than those treated with other commonly used ARBs [40]. The current guidelines state that only losartan, valsartan, and candesartan have effects comparable with ACE inhibitors in patients with advanced HF, but should only be used as a second-line treatment for patients intolerant to ACE inhibitors. In our study, treatment with fimasartan significantly increased zebrafish survival and functional status (Fig. 5). The in vivo results of this animal study suggested that human clinical studies are warranted to further evaluate the efficacy of fimasartan in patients with HF.
The limitation of this current study is that although zebrafish hearts have similar features to human hearts, they only have a single atrium and ventricle. Furthermore, our study evaluated terfenadine treatment-related HF for 4 days, and that a longer period of observation may be necessary to evaluate the full scope of HF.
Fimasartan prevented HF symptoms and modified physiologic changes in an in vivo zebrafish model of terfenadine-induced HF. Furthermore, fimasartan also decreased the number of apoptotic cells and increased zebrafish survival and motility. Human studies are warranted for further evaluation of the role of fimasartan in the treatment of HF.
KEY MESSAGE
1. Zebraf ish exposed to terfenadine showed changes similar to that of human heart failure (HF).
2. Treatment with fimasartan, an angiotensin receptor blocker, prevented symptoms and modified physiologic changes in an in vivo zebrafish model of HF.
3. Fimasartan could be a candidate drug for human HF, and further evaluations are warranted.


 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Supplement 1
Supplement 1 Print
Print





