


INTRODUCTION
Cytomegalovirus (CMV) is a major cause of morbidity and a preventable cause of mortality in immunocompromised patients [1]. CMV is frequently reactivated in immunocompromised patients, so where there is evidence of CMV replication it is difficult to differentiate CMV disease from bystander activation [2]. The serologic test of CMV, the detection of CMV viremia (i.e., CMV deoxyribonucleic academia by polymerase chain reaction [PCR], pp65 CMV antigenemia), and virus isolation by CMV culture or PCR from various specimen are the mainstay for the diagnosis of CMV infection. In addition, the direct examination of CMV-specific histopathologic findings with CMV immunohistochemistry or PCR in tissue biopsy is useful in the diagnosis of localized CMV tissue-invasive disease. However, the current diagnostic tests mainly based on viral load testing are suboptimal to diagnose or predict CMV disease differentiated from bystander activation. In the past 20 years, it has become clear that CMV-specific immunity plays a critical role in the development and severity of CMV disease [2]. Since the analysis of CMV-specific T cell response can potentially allow direct quantification of the patientŌĆÖs ability to control CMV, combining interpretation on host factors and viral factors such as CMV-specific T cell responses and data on CMV replication should help in this difficult clinical situation. Recently, interferon-╬│ releasing assays (IGRAs) for CMV have been developed. Two commercial CMV IGRA have become available on the writing of this manuscript; one is the enzyme-linked immunosorbent assay (ELISA)-based QuantiFERON-CMV (Cellestis, Valencia, CA, USA) and the other, the enzyme-linked immunospot assay (ELISPOT)-based T-track-CMV (Lophius Biosciences, Regensburg, Germany). Theoretically, combining observations on host factors and viral factors such as CMV-specific T cell responses and data on CMV replication should help in this difficult clinical situation. We report here the clinical application of such a combined interpretation in two different CMV co-infection models: (1) confirmed Pneumocystitis jirovecii pneumonia (PCP) patients with suspected CMV pneumonia and (2) patients with suspected gastrointestinal CMV disease.
METHODS
Study design and definitions
We prospectively enrolled all non-human immunodeficiency virus (HIV) infected adult patients with confirmed PCP combined with suspected CMV pneumonia, and those with suspected gastrointestinal CMV disease, who were admitted between January 2014 and December 2014 to the Asan Medical Center, a 2,700-bed tertiary hospital in Seoul, South Korea (Fig. 1). Tests for CMV immunoglobulin G were performed in the hematopoietic stem cell transplant (HCT) recipients and solid organ transplant recipients. All HCT or solid organ transplant recipients were monitored for CMV antigenemia during the post-transplant period. CMV antigenemia of a certain threshold (i.e., > 50 cells per 200,000 cells for kidney transplant and > 1 cells per 200,000 cells for HCT) were indications for preemptive ganciclovir therapy. Microbiological and pathological specimens for diagnosing CMV pneumonia and gastrointestinal CMV disease were processed using standard techniques and procedures, as described previously [3-6]. Decisions regarding anti-CMV therapy, such as ganciclovir, were made by the attending physicians based on each patientŌĆÖs initial clinical features, blood tests, microbiologic results, image findings, and histopathologic findings. To avoid biases, the results of the CMV IGRA were concealed from the attending physicians; this was because the results of the CMV IGRA could have affected decisions on empirical anti-viral therapy. The clinical categories of patients with suspected CMV pneumonia or CMV gastrointestinal (GI) diseases are summarized in Table 1 [3,4]. The study protocol was approved by the Institutional Review Board of Asan Medical Center (no. 2014-0198).
Microbiologic methods
PCP was diagnosed with a positive test result for an immunohistochemical (IHC) antibody assay for P. jirovecii using bronchoalveolar lavage (BAL) fluid in patients with respiratory symptoms and radiologic findings compatible with PCP. All the IHC tests for PCP were read by one experienced medical microbiologist (H.S.). BAL specimens were assayed routinely for CMV infection by shell vial culture. The CMV shell vial consisted of an MRC5 vial inoculated with 0.2 mL of cell specimen, centrifuged at 1,000 ├Śg for 15 minutes, incubated at 35┬░C overnight, and developed with the antibody in a DFA Cytomegalovirus Immediate Early Antigen Identification Kit (Diagnostic Hybrids, Athens, OH, USA). The CMV antigenemia assay using monoclonal antibodies C10/C11 (Biotest, Dreieich, Germany) was performed as previously described [3-6]. Counts were expressed as positive cells per 200,000 leukocytes. CMV DNA load (CMV quantitative PCR [qPCR]) in EDTA (ethylenediaminetetraacetic acid)-plasma and BAL was measured by qPCR a Cobas Amplicor CMV Monitor-Test Kit on a COBAS Amplicor Analyzer (Roche Molecular Systems, Branchburg, NJ, USA).
ELISPOT
A peripheral venous blood sample was collected from each patient for the CMV ELISPOT assay for interferon-╬│ producing T cell responses (i.e., T-track CMV). Briefly, peripheral venous blood (~8 mL) was obtained and peripheral blood mononuclear cells (PBMC) were immediately (within 30 minutes) separated and collected. The collected cells were suspended at a concentration of 2.0 ├Ś 106 cells/mL, placed (2.0 ├Ś 105 cells/well) in 4 wells pre-coated with anti-human interferon-╬│ antibody, and incubated for 18 hours. As a positive control PHA (Sigma-Aldrich, St. Louise, MO, USA) was added to other wells together with urea-formulated pp65 and urea-formulated IE1 as stimulation antigens, and medium alone served as a negative control, The resulting spots were counted with an automated microscope (ELiSpot 04 HR, Autoimmune Diagnostika GmbH, Strassberg, Germany) and the background value, obtained from the negative control wells, was subtracted.
Statistical analysis
This study is a preliminary proof-of-concept study, so no sample size was calculated. Our hypothesis was that infections with data located in the left upper quadrant (high viral load and low CMV-specific immunity) of the 2-axis model would be true CMV infections. We examined receiving operator characteristic (ROC) curves that plotted sensitivity against the rate of false-positive results over a range of cut-off values. We chose the optimal cut-off value as the point on each ROC curve farthest from the diagonal line that maximized the sum of sensitivity and specificity. Diagnostic performance was expressed in terms of sensitivity, specificity, positive predictive value, and negative predictive value. However, each cut-off value for CMV PCR and CMV IGRA, respectively, in the 2-axis model was selected to maximize the diagnostic performance.
RESULTS
Patients characteristics
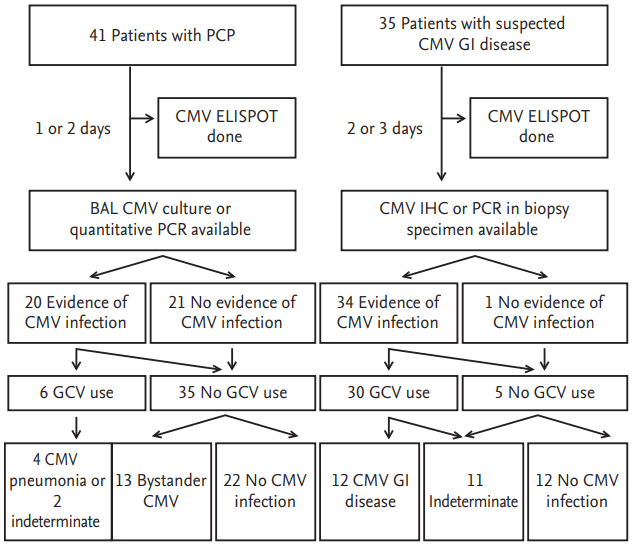
During the study period, 41 patients with PCP and 35 patients with suspected CMV GI disease were enrolled (Fig. 1). The baseline clinical characteristics of the patients with PCP and with suspected CMV GI disease are shown in Tables 2 and 3, respectively. Of the 41 patients with PCP, four (10%) were classified as combined CMV pneumonia, 13 (32%) as bystander activation, and 22 (56%) as no CMV infection. The remaining two (5%) were classified as indeterminate combined CMV pneumonia and excluded from the final analysis. Of the 35 patients with suspected CMV GI disease, 12 (34%) were classified as confirmed CMV GI disease and 12 (34%) as no CMV GI disease with bystander activation. The remaining 11 (31%) were classified as indeterminate CMV GI disease and excluded from the final analysis. Finally, the 39 patients with PCP and the 24 patients with CMV GI disease and no CMV GI disease were included in our analysis of the proposed model.
2-Axis model in the patients with PCP
All the 39 patients with PCP were diagnosed on the basis of positive immunohistochemical staining for PCP and received trimethoprim-sulfamethoxazole as primary drug. The detailed microbiologic results for CMV are shown in Table 2. The sensitivity, specificity, and area under ROC curve of CMV IGRA, BAL CMV qPCR, blood CMV qPCR, and CMV antigenemia for diagnosing combined CMV pneumonia in patients with PCP were shown in Table 4. The area under the ROC curves of pp65-specific T cell response for CMV diseases in patients with PCP was 55 (95% confidence interval [CI], 39 to 71). Fig. 2 is a dot plot showing the position of each patient in the 2-axis model where the x-axis gives the CMV IGRA results and y-axis the results of the BAL CMV qPCR. It can be seen that the dots representing all four patients with PCP and CMV pneumonia are in the left upper quadrant of the 2-axis model, as predicted. The sensitivity, specificity, positive predictive value, and negative predictive value of high BAL CMV titer (more than 3.5 log) and low CMV IGRA result (less than 50 spot forming cells (sfc)/200,000 PBMC) for diagnosing combined CMV pneumonia in patients with PCP were 100% (95% CI, 40 to 100), 100% (95% CI, 90 to 100), 100% (95% CI, 40 to 100), and 100% (95% CI, 90 to 100), respectively. Other plots where the x-axis gives the CMV IGRA results and the y-axis the results for CMV virus replication as shown by BAL CMV culture, blood CMV qPCR or CMV antigenemia are presented in Supplementary Fig. 1. The diagnostic performance of BAL CMV qPCR and the CMV-specific ELISPOT for predicting 30-day mortality in PCP is shown in Supplementary Table 1.
2-Axis model in the patients with suspected CMV GI disease
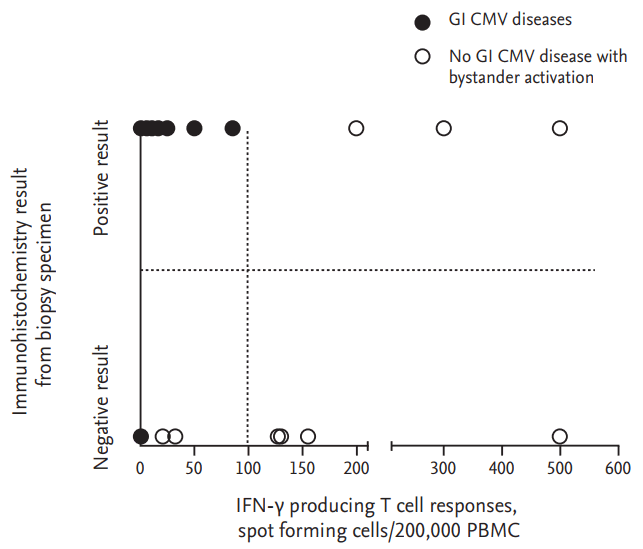
Of the 24 patients with suspected GI CMV disease, 12 were confirmed by positive immunohistochemical staining for CMV and received intravenous ganciclovir as primary drug. The detailed microbiologic results for CMV are shown in Table 3. The area under the ROC curves of pp65-specific T cell response for CMV diseases in patients with suspected CMV GI disease was 71 (95% CI, 53 to 85). Fig. 3 is a dot plot for each patient in the 2-axis model where the x-axis is the CMV IGRA results and the y-axis the IHC result for CMV in biopsy tissue. As can be seen, 11 (92%) of the cases fell, as expected, in the left upper quadrant of the 2-axis model. The sensitivity, specificity, positive predictive value, and negative predictive value of CMV replication in biopsy tissue (positive IHC staining) and low CMV IGRA result (100 sfc/200,000 PBMC) for predicting GI CMV disease in patients with suspected GI CMV disease were 92% (95% CI, 62 to 100), 100% (95% CI, 74 to 100), 92% (95% CI, 62 to 99), and 100% (95% CI, 73 to 100), respectively. Other models where the x-axis gives the CMV IGRA results and the y-axis the results for CMV viral replication, as shown by CMV PCR in biopsy tissue or blood CMV antigenemia, are presented in Supplementary Fig. 2.
Association between clinical disease category and responses to IE1 and pp65
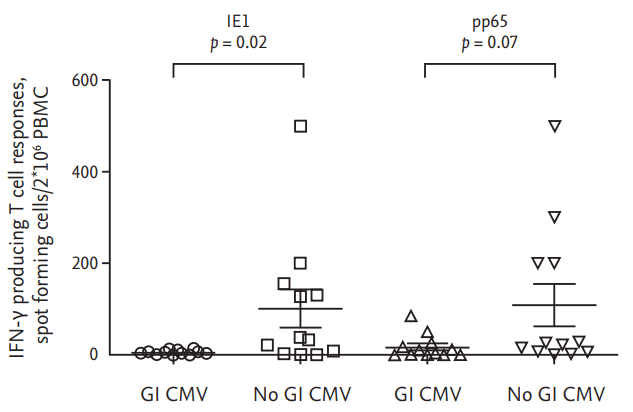
We investigated whether the responses of the 24 patients with suspected GI CMV disease to IE1 and pp65 were associated with their clinical category (Fig. 4). There was a significant association between clinical disease category and responsiveness to IE1 (p = 0.02), and there was a non-significant indication of an association between responsiveness to pp65 and clinical category (p = 0.07).
DISCUSSION
CMV is frequently reactivated in immunocompromised patients, so when there is evidence of CMV replication it is difficult to differentiate true CMV infection from bystander activation. Until now, diagnosis of CMV disease depended on quantitative or qualitative data on CMV replication in blood, tissue, or urine, together with non-quantitative host factors such as transplant recipient, steroid user, or immunosuppression. However, the presence of a given host risk factor is not merely qualitative, but also a crude measure of host immunologic susceptibility to a certain pathogen such as CMV. The recently developed IGRAs for CMV make it possible to quantify individual immunologic susceptibility to CMV. Theoretically, the combined interpretation of host factors and viral factors such as CMV-specific T cell responses with the data on CMV replication would more accurately differentiate CMV disease from bystander activation. This proof-of-concept study showed that the data for true CMV infections fell mostly into the left upper quadrant of the proposed 2-axis model corresponding to infections giving a low CMV-specific cell-mediated immune response along with evidence of (high) virus replication at the local infection site. Hence, our proposed 2-axis model appears to be useful for differentiating true CMV infection from bystander activation.
It has been reported that CMV replication is frequently observed (20% to 70% positive BAL CMV cultures) in HIV-infected patients with PCP [7]. In general, treatment of the PCP alone often results in clinical improvement without the need to treat the CMV [8,9]. Therefore, most of the positive BAL CMV cultures in HIV-infected patients with PCP are regarded as the result of bystander activation. We previously found that about one-thirds of non-HIV patients with confirmed PCP give positive BAL CMV culture results [10]. However, it is largely unknown what proportion of such patients have true coinfections rather than bystander activation in non-HIV patients with PCP. In this study, we differentiated true CMV co-infection from bystander activation according to whether clinical improvement occurred with/without ganciclovir. About half of the patients with PCP (19/41) showed evidence of CMV replication, but only four (10%) were classified as CMV pneumonia. Furthermore, our proposed 2-axis model differentiated exactly between true CMV infection and bystander activation. It would therefore be worth investigating whether the proposed 2-axis model also works in HIV-infected patients with PCP. Since the pathophysiologic basis of CMV pneumonia is different in HIV-infected patients and non-HIV patients, different models may be needed to differentiate CMV disease from bystander activation in these two cases. A large prospective study is needed to confirm our conclusion that the combined interpretation of CMV-specific T cell response and CMV replication is of help in diagnosing and treating CMV pneumonia.
GI CMV disease is a major cause of morbidity and mortality in immunocompromised patients, especially transplant recipients [1] and occasionally causes severe complications in immunocompetent hosts, especially in those of advanced age [11]. The identification of characteristic intranuclear inclusions in stained tissue where the presence of CMV is suspected has been used as the standard reference method for confirming CMV GI disease [4]. However, positive results for immunohistochemical staining or CMV PCR of a biopsy specimen may not be sufficient to diagnose CMV GI disease, although these combined diagnostic method do increase the sensitivity of detection of CMV GI disease. That is because it is difficult to differentiate CMV disease from bystander activation in patients with suspected CMV colitis by using only qualitative evidence of CMV replication at a local site, such as positive IHC or CMV PCR. Our data suggest that the presence of a low CMV-specific T cell response as part of the evidence for CMV replication at a local site can help in differentiating CMV GI disease from bystander activation in this difficult clinical situation.
Immunosuppressive therapy in inflammatory bowel disease (IBD) frequently induces CMV reactivation in the colonic mucosa, and in turn exacerbates IBD [12,13]. It is often difficult to accurately differentiate CMV GI disease from bystander activation in patients with IBD, especially ulcerative colitis [14]. Actually, most patients with IBD with evidence of colonic CMV received ganciclovir therapy in our cohort, as specified in the guidelines of the American College of Gastroenterology [15]. However, the detection of CMV in the colon of a patient with IBD does not necessarily mean GI CMV disease because some patients with CMV detected in their colonic mucosa and not treated with antivirals also go into remission [16]. Therefore, most such cases, if they received ganciclovir therapy, were classified as indeterminate CMV GI disease in this study. Interestingly, high CMV-specific T cell responses were seen in about half such patients (Supplementary Fig. 2C). It is possible that infections in some patients with high CMV-specific T cell responses resolve spontaneously without use of an antiviral agent, or that they are less resistant to immunosuppressive therapy. In addition, there are promising results showing that CMV GI disease in the inflamed mucosa of patients with ulcerative colitis can be differentiated from bystander activation by quantitative real-time PCR [16,17]. Therefore, it would be interesting to evaluate the usefulness for guiding ganciclovir treatment of combined interpretation of quantitative CMV-specific T cell responses and quantitative evidence of CMV replication in the colonic mucosa by differentiating between CMV GI disease and bystander activation or predicting resistance to immunosuppressive therapy. Furthermore, the detailed kinetic data about CMV-specific T cell response before and after antiviral therapy will give us more insight on the understanding of pathophysiology of CMV diseases.
Both pp65 and IE1 are considered major T cell targets [13]. A previous study demonstrated that pp65-specific immunity was crucial for controlling the dissemination of CMV in an animal model [18] and that there was a positive correlation between pp65 T cell responses and CMV viremia [19]. In contrast, others have shown that the level of IE1-speicific T cells correlated with protection from CMV disease in solid organ transplant recipients [17] and was useful for predicting the post transplant risk of CMV infection in kidney transplant recipients [20]. Therefore, our findings of higher levels if IE1-specific T cells than pp65-specific T cell in association with no GI CMV disease is consistent with these previous studies [17,20].
This study has several limitations. First, it is a proof-of-concept study and the sample size was not enough to calculate the diagnostic performance of the 2-axis model for CMV disease. Second, there is no established gold standard test for the diagnosis of CMV disease; hence our cases could not be firmly classified as CMV disease. To avoid biases, the results of the CMV IGRA were concealed from the attending physicians because the results of the CMV IGRA could have affected decisions on empirical anti-viral therapy. The classification of bystander activation was relatively clear because clinical improvement without antiviral treatment could be convincingly classified as due to it. However, some patients in which bystander activation actually occurred may have been classified as CMV disease or indeterminate CMV disease if they received antiviral treatment. Finally, our study was not appropriately designed to evaluate the diagnostic performance of CMV quantitative PCR because this test was integrated in the diagnostic criteria. So, the cautious interpretation needed because of a bias favoring diagnostic performance of CMV quantitative PCR.
In conclusion, this study suggests that infections with low CMV IGRA results and high levels of CMV virus replication are true CMV infections. This concept might help clinicians make decisions about antiviral therapy, so further studies with large number of cases in different types of CMV diseases should be proposed.
KEY MESSAGE
1. Cytomegalovirus (CMV) is frequently reactivated in immunocompromised patients, so it is difficult to differentiate true CMV infection from bystander activation when there is evidence of CMV replication.
2. We created the 2-axis model with the CMV interferon-╬│ releasing assay results as the x-axis and the results for CMV virus replication as the y-axis.
3. Cases falling in the left upper quadrant (high viral load and low CMV-specific immunity) of the model appear to be true CMV infections.

 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Supplement 1
Supplement 1 Print
Print





