


INTRODUCTION
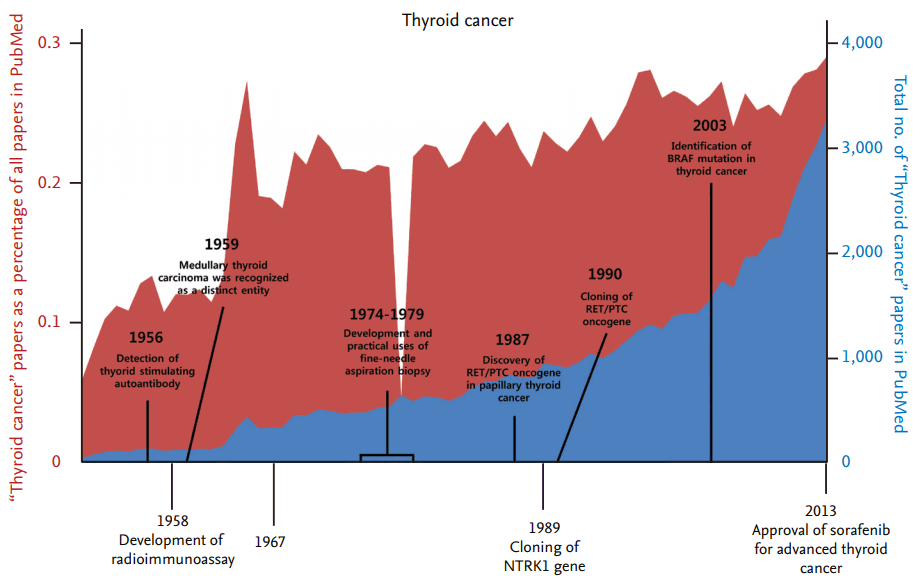
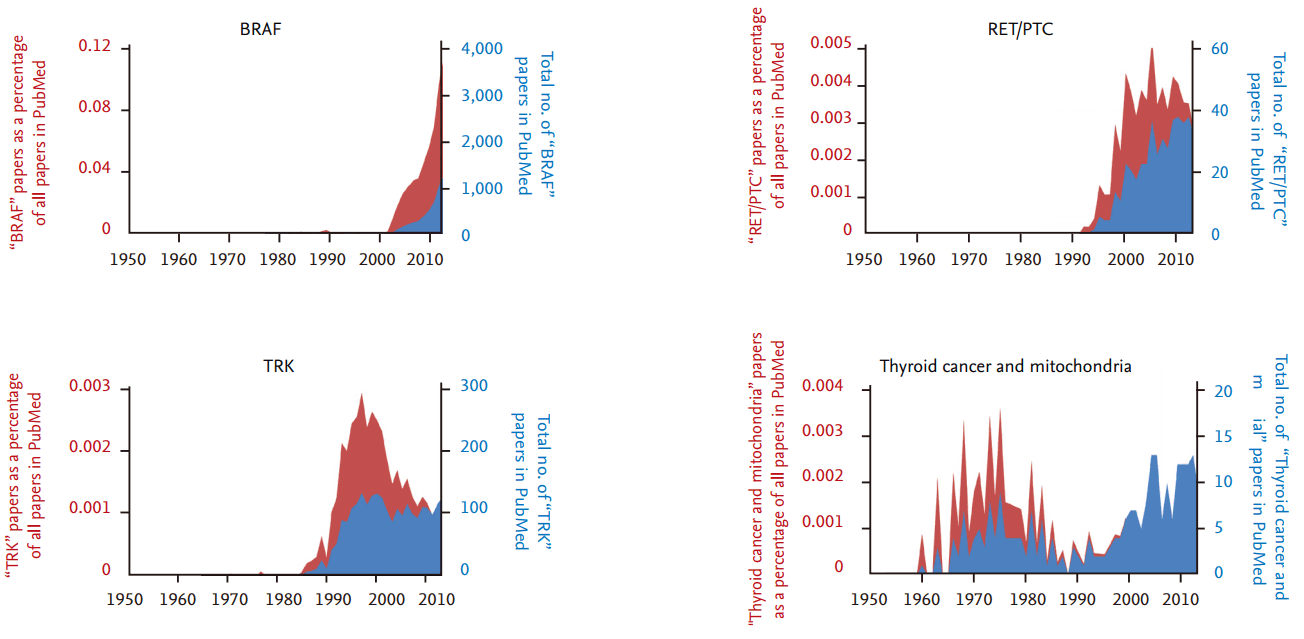
Thyroid cancer is one of the most common malignancies of endocrine organs, and its incidence rate has steadily increased over the past several decades [1]. From the late 1940s on, the number of research articles on thyroid cancer has continuously increased, although there has been no significant difference in the number of thyroid cancer articles as a percentage of all research articles (Fig. 1). From the late 1980s to 2000, the discovery and functional validation of oncogenes stimulated an increase in articles related to several important oncogenes that also play a critical role in thyroid tumorigenesis (Fig. 2). The number of articles exploring the relationship between thyroid cancer and mitochondria increased during the 1970s, and has increased again since the early 2000s.
The thyroid gland contains two distinct types of epithelial cells, designated as follicular and parafollicular cells. Although both cell types may develop into distinct cancers, more than 95% of thyroid cancers originate from follicular epithelial cells. Most differentiated thyroid tumors derived from thyroid epithelial cells exhibit slow-growing cancers, and patients with these tumors can achieve a good prognosis with surgical removal and radioiodine treatment [1].
The recent progress in understanding the molecular mechanisms of thyroid cancers has promoted the development of effective treatment modalities [2]. This has resulted from the discovery of molecular events including germline or somatic mutations, genetic rearrangements, and epigenetic alterations that constitutively stimulate signaling pathways such as the mitogen-activated protein kinase (MAPK) and phosphatidylinositol-3-kinase (PI3K)-Akt pathways [2]. Tumorigenesis is a complex process that is regulated by the activation of oncogenes, inactivation of tumor suppressors, and reprogramming of cell death [3]. Mitochondria also play an essential role in tumorigenesis as vital organelles responsible for energy production, generation of radical oxygen species (ROS), calcium homeostasis, and the regulation of apoptotic signaling [4]. Cancer cell proliferation and resistance is regulated by these mitochondrial properties during the development of cancer. Mitochondrial energy metabolism is implicated in the alteration of energy production in tumor cells. Additionally, recent studies show that mitochondrial quality control through autophagy and mitophagy is associated with the growth of tumors, including thyroid cancer tumors [5,6].
This review focuses on the recent remarkable progress in understanding the molecular mechanisms of thyroid cancer, and discusses the role of altered mitochondrial metabolism in the development of thyroid cancer.
GENETIC ALTERATIONS IN THYROID CANCER
Genomic mutations or rearrangements regulating growth-stimulating signaling pathways appear to be involved in the transformation process in well-differentiated thyroid cancers [7,8]. A prominent mutation is the V600E point mutation of B-Raf (BRAF), which constitutively activates this serine/threonine kinase [9]. BRAFV600E mutations in papillary thyroid cancer (PTC) cell lines are indispensable for tumor initiation, proliferation, and tumorigenicity [10]. BRAFV600E transgenic mice also develop a more aggressive phenotype through the regulation of the MST1 (macrophage stimulating 1)-FOXO3 (forkhead box O3) signaling pathway in PTC [11]. One study showed that the BRAFV600E mutation is detected in nearly 45% of PTCs [12]. The BRAFV600E mutation is strongly associated with poor clinicopathologic outcomes including mortality, lymph node metastasis, and distant tumor metastasis among patients with PTC [13]. Moreover, other studies also support the effect of the BRAFV600E mutation on poor prognosis in patients with PTC [14]. However, overall mortality is very low in PTC and questions still remain as to how best to utilize information regarding the BRAFV600E mutation in managing mortality risk.
Ras mutations are also prevalent in thyroid cancer. Ras is a member of the G protein families, which bind GDP (guanosine diphosphate, inactive form). or GDP (guanosine diphosphate, inactive form). Mutations are known to induce the loss of Ras GTPase activity; thereby, locking Ras in a constitutively active GTP-bound form. Of the three mammalian Ras genes (HRas, KRas, and NRas), NRas is predominantly mutated in thyroid cancers, mostly showing mutations in codons 12 and 61. Ras mutations activate the MAPK and PI3K-Akt signaling pathways in thyroid tumorigenesis [15]. Ras mutations are also common in follicular thyroid adenoma, a premalignant lesion, indicating a role in early follicular thyroid cell tumorigenesis [16]. However, the HRas mutation is found in normal thyroid cells in vitro, and is associated with well-demarcated and differentiated colonies with phenotypes consistent with follicular thyroid adenoma and cellular proliferation with normal differentiation [17].
Balanced inversions or translocations resulting in oncogenic rearrangement, which usually involve the 3.0 kB intron 11 of the tyrosine kinase receptor protein RET, are found in thyroid cancer. In other words, RET-PTC rearrangements occur as a result of genetic recombination between the 3’ tyrosine kinase portion of RET and the 5’ portion of a partner gene. RET-PTC reportedly results in ligand-independent dimerization and constitutive tyrosine kinase activity in thyroid epithelial cells. Although RET-PTC has intrinsic tyrosine activity and occurs in thyroid tumors, the direct substrate of RET-PTC in thyroid cells remains unknown. Previously, our group showed that phosphoinositide-dependent kinase 1 (PDK1), an essential serine/threonine kinase in growth factor signaling pathways, is a target of RET/PTC. RET-PTC and PDK1 are colocalized in the cytosol, and RET-PTC phosphorylates a specific tyrosine (Y9) residue located in the N-terminal portion of PDK1. RET-PTC-induced tyrosine phosphorylation of PDK1 is one of the mechanisms by which it acts as an oncogenic tyrosine kinase in thyroid tumorigenesis [18]. Additionally, RET-PTC also phosphorylates a Y315 residue in protein kinase B (PKB), which is known to be phosphorylated by the Src tyrosine kinase. This event activates PKB downstream signaling pathways; thereby, inducing thyroid carcinogenesis [19].
RET-PTC also interacts with signal transducer and activator of transcription 3 (STAT3), and phosphorylation activity of RET-PTC on a specific residue (Y705) of STAT3 is a critical signaling event for the induction of genes in the RET-PTC-mediated transformation in thyroid carcinogenesis in vitro [20]. An immunoprecipitation experiment showed an interaction between liver kinase B1 (LKB1) and STAT3. Moreover, LKB1 and activated STAT3 were colocalized within the nucleus of the cell. Furthermore, LKB1 reduces STAT3 transcriptional activity in RET-PTC transfected cells. Thus, we suggested that LKB1 suppresses tumor growth as a transcriptional co-repressor of oncogenic STAT3, which is activated by RET-PTC [21]. Additionally, the expression of phospholipase D is increased in papillary thyroid carcinoma. Transcriptional activity of STAT3 induced by RET-PTC is enhanced by overexpression of phospholipase D; thereby, indicating that phospholipase D plays a synergistic role in regulating the RET-PTC-induced STAT3 activation during thyroid carcinogenesis [22]. Thyroid stimulating hormone (TSH)-mediated STAT3 activation is associated with SOCS (suppressor of cytokine signaling)-1 and -3 induction, which play a role in the resistance of the thyroid cells to interferon γ during the thyrocyte growth phase [23]. TSH receptor-activated PI3K signaling induced by S6K1 (S6 kinase beta-1) modulates thyrocyte proliferation and thyroid follicle activity [24]. Moreover, in a recent study, SU11248, which is a multi-target tyrosine kinase inhibitor, induced a complete morphological reversion of transformed NIH-RET/PTC3 cells and inhibited the growth of TPC-1 (two pore segment channel 1) cells that have endogenous RET-PTC1 [25]. RET-PTC can also phosphorylate and activate STAT1. This may lead to increased expression of major histocompatibility complex (MHC) class II, which may explain why the tissues surrounding RET-PTC-positive cancers are infiltrated with lymphocytes [26].
Other important genes that are responsible for thyroid tumorigenesis include p53, phosphatase and tensin homolog (PTEN), β-catenin, isocitrate dehydrogenase 1, anaplastic lymphoma kinase, and epidermal growth factor receptor [27-32]. The paired box 8-peroxisome proliferator-activated receptor γ (PPARγ) fusion gene is also another recombinant oncogene in follicular thyroid carcinoma [33]. Moreover, the AKAP9 (A-kinase anchor protein 9)-BRAF fusion gene results in an increase in BRAF kinase activity, associated with ionizing radiation-induced PTC, but not with sporadic PTC [34]. Oncogenic kinases involved in papillary thyroid carcinoma trigger the expression of class II transactivator and MHC class II complex [35]. Additionally, the expression of the Gadd45γ (growth arrest and DNA damage-inducible gene family 45 γ) gene, which has been implicated in a variety of growth-regulatory mechanisms, is significantly decreased in anaplastic thyroid cancer cell lines. Re-expression of Gadd45γ suppressed anaplastic thyroid cancer cell growth, reminiscent of the effects of p53 [36]. Acquisition of functional CXCR4 (C-X-C chemokine receptor type 4) expression is involved in local invasion by autocrine and paracrine SDF-1α (stromal cell-derived factor-1α) stimulation during the process of carcinogenesis in anaplastic thyroid cancer [37]. Although most of these genetic alterations are common in thyroid cancer, genetic alterations have not been characterized in approximately 30% of differentiated thyroid carcinomas. Therefore, further studies are necessary for determining the underlying genetic mechanisms of thyroid tumorigenesis.
MITOCHONDRIAL METABOLISM IN TUMORIGENESIS
The energy metabolism of most cancer cells differs markedly from that of normal cells in order to meet energy demands during tumor progression [38]. It has become clear that cellular energy metabolism controlled by oncogenes and other tumor-related factors is a critical factor determining the clinical phenotypes of cancer. Our understanding of the molecular carcinogenesis of thyroid cancer has notably advanced over the last decade. However, the role and nature of energy metabolism in thyroid cancer remains to be identified.
Mitochondria provide 90% of the cellular energy required for various biological functions through oxidative phosphorylation in the inner mitochondrial membrane [39]. In addition, mitochondria regulate various cellular processes, including steroid hormone and porphyrin synthesis, the urea cycle, lipid metabolism, and the interconversion of amino acids [40]. Mitochondria also play central roles in apoptosis, cellular proliferation, and cellular Ca2+ homeostasis, which affect numerous cellular signaling pathways. Taken together, mitochondria play an important role in the energy metabolism of the normal thyroid gland as well as in thyroid tumors. The consequences of mitochondrial functional and structural alterations in thyroid tumorigenesis and tumor progression will be further explored in the next section.
Mitochondrial DNA alterations in thyroid cancer
The genetic basis of cancer progression has already been studied extensively and will not be covered in depth in this review. Increases in the somatic or germline mutation frequency induced by DNA repair defects, replication errors, chemical or radiation exposure, or the aging process have been reported to lead to cancer [41]. In contrast, the role of mitochondrial genomic mutations in the development of thyroid cancer has not been well studied. The majority of mitochondrial proteins are encoded by nuclear DNA, with only 37 genes encoded by mitochondrial DNA (mtDNA). These include 13 components of the electron transport chain, 22 transfer RNAs, and two ribosomal RNAs. MtDNA mutations influencing mitochondrial metabolism and biogenesis have been found in a variety of cancers [42-47]. Several studies have shown that acquired mtDNA alterations are associated with thyroid cancer. It has been known for over three decades that thyroid tumors contain abnormally high numbers of mitochondria. A common mutational mechanism in thyroid cancer is somatic point mutations of mtDNA. The majority of the mutations were found in mitochondrial complex I of the respiratory chain, and severe functional defects in complex I activity were observed in thyroid tumor cell lines [48]. These studies suggest that somatic mtDNA mutations may be associated with tumor development in the thyroid gland. Disruptive mtDNA mutations in complex I of the respiratory chain are also markers for oncocytic thyroid tumors and may promote the accumulation of abnormal mitochondria [49]. Moreover, mitochondrial dysfunction in the oncocytic thyroid cell line XTC.UC1 can be attributed to combined complex I/III defects associated with mtDNA mutations [50]. A study of 79 benign and malignant thyroid tumors revealed that mtDNA deletions were detected in 100% of Hürthle cell tumors, 33.3% of adenomas, and in 18.8% of non-Hürthle cell papillary carcinomas [51]. Additionally, the number of mtDNA deletions was strongly correlated with the level of mtDNA content in tumor tissues in radiation-related human PTC and follicular adenoma [52].
Mitochondrial biogenesis and metabolism in thyroid cancer
Mitochondrial biogenesis is a complex and multistep cellular process resulting in the addition of new mitochondrial material within a cell. Emerging evidence shows that it is also associated with carcinogenesis in thyroid cancer as well as other types of tumors. Oncocytomas are defined by an abnormal proliferation of mitochondria, and have an effect on a variety of human tissues. Thyroid oncocytomas, also known as Hürthle cell tumors, are characterized by the presence of abundant oxyphilic granular cytoplasm in at least 75% of the oncocytes due to the accumulation of numerous abnormal mitochondria [53,54]. Oncocytic changes in thyroid follicular cells are frequently detected in obese mice and humans, and abnormal mitochondria have been observed in the thyroids of obese mice [55]. Mitochondrial biogenesis is regulated by nitric oxide (NO) and calcium via PGC-1-related coactivator (PRC) in human follicular thyroid carcinoma cell lines [56]. NO also controls mitochondrial biogenesis through the cGMP (cyclic guanosine monophosphate)/PRC pathway in a cellular model of oncocytic thyroid tumors [57]. Mitochondrial dynamics are indispensable for thyroid oncocytic cell tumors and play an important role in the development of the malignant phenotype. Proteins governing mitochondrial dynamics, such as mitofusin 2 (MFN2), optic atrophy 1 (OPA1), dynamin-related protein 1 (DRP1), and mitochondrial fission 1 (FIS1), are overexpressed in oncocytic cell tumors [58]. Among these proteins, the expression of Mfn2 and the pro-fission protein Drp1 were found to be positively associated with malignant oncocytic thyroid tumors [58]. Furthermore, a blockade of Drp1 is sufficient to decrease oncocytic cell migration and invasion [58].
Mitochondrial function in oncogenic action in thyroid cancer
Molecular events associated with the development of thyroid cancer include activation of the RET/PTC and TrkA (tropomyosin receptor kinase A) tyrosine kinase receptors, activation of Ras genes, inactivation of the p53 gene, Pax8, and PPARγ1 rearrangements, and the active conformation of BRAFV600E [2]. The activation of the RET/PTC oncogene is prevalent in Hürthle cell thyroid adenomas and carcinomas [59]. RET/PTC rearrangement-induced STAT3 activation is also frequently observed in cancer cells [20] and has been implicated as a regulator of cellular metabolism in tumor formation [60]. Moreover, STAT3 phosphorylated at serine 727 has been shown to localize in the mitochondria, where it positively regulates the activity of mitochondrial complex I/II [61]. LKB1 suppresses RET/PTC-dependent activation of STAT3 and represses the binding of STAT3 to its target promoters; thereby, decreasing the expression of downstream target genes of STAT3 in a human thyroid cancer cell line [21]. However, the tumor-promoting role of STAT3 has been challenged by several studies [62-64]. STAT3 knockdown in a thyroid cancer cell line and a murine model of BRAFV600E-induced PTC increased cellular proliferation and tumor growth [65]. The mitochondrial localization observed in oncogenic BRAF mutants may be related to the altered responses to apoptotic stimuli and characteristic metabolic phenotypes found in thyroid cancer [66].
The presence of activated mutant Ras in thyroid tumors, including benign follicular adenomas, follicular carcinomas, and papillary carcinoma, has been known for several decades [67]. Ras proteins play an essential role in the activation of the MAPK and PI3K-Akt signaling pathways in thyroid cancer, which mediate cellular proliferation and survival [68,69]. Although Ras is a dual activator of both the MAPK and PI3K-Akt signaling pathways, Ras mutations primarily activate the PI3K-Akt pathway in thyroid tumorigenesis [15,70]. Activation of PI3K-Akt induces aberrant methylation and, thus, silencing of PTEN; thereby, leading to a failure to suppress PI3K-Akt signaling and thyroid tumor progression [71]. The Ras oncoprotein is also involved in altering mitochondrial function, genes, and ROS production [72,73]. K-Ras overexpression induces a rapid reduction of complex I and a decrease in mitochondrial membrane potential; thereby, leading to a switch from oxidative phosphorylation to glycolysis, which promotes tumorigenesis [74]. Moreover, mitochondrial STAT3 seems to contribute to Ras-dependent transformation by increasing electron transport chain activity [61].
The most frequently mutated tumor suppressor gene in human cancers is p53. It rapidly translocates to the mitochondrial outer membrane during p53-dependent apoptosis, which is associated with changes in mitochondrial membrane potential, cytochrome C release, and caspase activation [75]. In contrast, mitochondrial respiratory dysfunction also alters p53 expression and transcriptional activity, which consequently impairs p53-mediated cell death [76]. Impairment of p53 caused by germline variants in mitochondrial complex II-succinate dehydrogenase enhances the risk of thyroid cancer in Cowden and Cowden-like syndromes by altering FAD/NAD (flavin adenine dinucleotide/nicotinamide adenine dinucleotide) levels, which are linked to the destabilization of p53 [77].
Mitochondrial quality control and mitophagy in thyroid cancer
Biogenesis and mitophagy work in concert to regulate mitochondrial mass, function, and quality. Mitophagy is a specialized form of autophagy that selectively degrades and eliminates superfluous or damaged mitochondria.
Oncocytes (Hürthle cells or Askanazy cells) are recognized as a subset of cells characterized by an abundant cytoplasm in which functionally defective mitochondria aberrantly accumulate. Researchers recently found alterations in mtDNA that may perturb oxidative phosphorylation, and speculated about a compensatory organelle biogenesis [51]. It is conceivable that mitophagy is critically linked with the development of oncocytes, a prominent feature of Hürthle cell tumors in the human thyroid gland. We have previously reported that the induction of autophagy and autophagosome formation are common features of oncocytes in Hürthle cell tumors. However, the accumulation of defective mitochondria in oncocytes may be caused by the inability of mitophagy to sufficiently remove abnormal mitochondria.
The PTEN-induced putative kinase 1 (PINK1)-Parkin pathway plays a critical role in the maintenance of mitochondrial quality control by triggering the mitophagy of abnormal mitochondria [5,6]. Studies performed in the Hürthle cell-derived XTC.UC1 cell line showed that effective mitochondrial localization of Parkin in response to carbonyl cyanide m-chlorophenylhydrazone (CCCP) treatment was impaired, indicating inefficient mitophagy. These results suggest that XTC.UC1 cells have an intact process of non-selective autophagy, but may have defects in mitophagy regulated by PINK1 and Parkin. Low immunoexpression of PINK1 and Parkin was detected in Hürthle cell tumors. Surprisingly, in XTC.UC1 cells, Parkin failed to efficiently translocate into mitochondria following CCCP treatment, which is a critical molecular step in PINK1-mediated mitophagy. Based on these findings, the ineffective turnover of abnormal mitochondria in XTC.UC1 cells may be caused by inefficient mitochondrial translocation of Parkin, associated with decreased ligase activity of mutant Parkin in mitochondria [78].
In sum, oncocytes found in Hürthle cell tumors showed ineffective turnover of abnormal mitochondria that may be due to decreased E3 ligase activity associated with dysfunctional translocation of Parkin into mitochondria. In addition, some patients with Hürthle cell tumors harbored a tumor-specific mutation in Parkin (V380L), and this may explain why oncocytes form in a specific group of heterogeneous Hürthle cell tumors [78].
CONCLUSIONS
The current challenges lie in identifying the mechanisms modifying oncogenic potentials, which are driven by oncogenic factors, particularly those that mediate transformation, progression, and invasion. Mitochondrial energy metabolism is implicated in the alteration of energy production in tumors. Moreover, active mitochondrial remodeling and adaptation in tumor cells may control the tumor microenvironment and also affect molecular characteristics of the heterogeneity of tumor cells. Additionally, in recent studies, mitochondrial quality control by autophagy and mitophagy seems to be associated with tumor growth, including thyroid cancer [78]. These issues must be addressed for the effective control of advanced thyroid cancer, which has no effective treatment modalities at present.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print





