


INTRODUCTION
The 5-year survival rate of cancer patients in Korea has increased by 68.1%; consequently, the estimated number of patients who underwent cancer treatment was up to 1.37 million people in 2013 [1]. The number of patients receiving chemotherapy and surviving after chemotherapy is also gradually increasing. Caution is required for most anti-cancer agents currently in use due to various adverse effects, and cardiotoxicity is a well-known major adverse effect that impacts the quality of life and mortality of cancer patients [2]. The development of cardiotoxicity during chemotherapy limits the proper treatment of cancer itself and thus deteriorates the quality of life if it results in overt heart failure.
Cardiotoxicity can increase mortality not only due to cancer itself but also due to heart failure even if the patient has survived cancer. Many types of cardiotoxicity have been reported, including bradycardia, QT interval prolongation, myocardial ischemia, and cardiomyopathy [3], and one type of anti-cancer drug may cause various types of cardiotoxicity [2-4]. The pathogenesis of cardiotoxicity is different for each drug. Additionally, the pathogenesis of anthracycline-induced cardiotoxicity has not yet been clearly determined; this lack of knowledge is a major obstacle to the prevention and treatment of cardiotoxicity.
Although the anthracyclines are representative chemotherapeutic agents that can cause cardiotoxicity, particularly left ventricular dysfunction, they are very effective chemotherapeutic agents for use in treating breast cancer, lymphoma, leukemia, and sarcoma, among others. The early generation anthracycline compounds daunorubicin and doxorubicin can cause fatal heart failure [5]. Anthracyclines developed later, such as idarubicin, epirubicin, and mitoxantron, are less cardiotoxic than the earlier generation drugs, but cardiotoxicity is still a problem. Doxorubicin, an anthracycline compound, has been studied extensively, and the incidence of cardiotoxicity increases according to the cumulative dose [6]. The development of subclinical cardiotoxicity at a cumulative doxorubicin dose less than 300 mg/m2 was recently reported [7].
There has been tremendous effort to understand the pathophysiology of anthracycline-induced cardiotoxicity and research to find an appropriate treatment, but no effective preventive drug has been developed for use in clinical practice. Because anthracycline-induced cardiotoxicity can cause irreversible damage to the myocardium [6], the early detection of cardiotoxicity and prevention of progression to heart failure is the most effective strategy [7-9]. In clinical practice, each cancer type has different clinical characteristics, dosing schedules, and combination regimens; therefore, it is difficult to suggest a recommendation that can be applied to any cancer type. The current understanding of the pathophysiology of anthracycline-induced cardiotoxicity and awareness of monitoring, prevention, and treatment strategies are important in the management of patients receiving anthracyclines.
DEFINITION OF CARDIOTOXICITY
The most commonly accepted definition of cardiotoxicity in the oncology community is an absolute decrease of 10% or more in the left ventricular ejection fraction (LVEF) from baseline or an LVEF decline to < 50% compared with the baseline [10]. However, some studies were conducted with a normal cut-off value of 55% of LVEF, as measured by echocardiography [11,12]. Cardiotoxicity is also defined as a LVEF decline of Ōēź 5% to < 55% with heart failure symptoms or an asymptomatic decrease in LVEF of Ōēź 10% to < 55% in patients treated with trastuzumab, a monoclonal antibody for the treatment of breast cancer [13]. Therefore, physicians may be confused as there is not yet a consensus definition for cardiotoxicity that can be applied to all cancer types.
There are also imaging modality discrepancies in the measurement of LVEF. Multigated radionuclide angiography (MUGA) is reliable and was commonly used for the evaluation of LVEF in anthracycline-induced cardiotoxicity before two-dimensional (2D) echocardiography became the gold-standard method for measuring LVEF. A comparative study demonstrated that the average LVEF measured by MUGA was less than that of echocardiography in the same group of patients [14], suggesting that the choice of imaging modality can affect the detection of cardiotoxicity. It is reasonable to define cardiotoxicity by the consensus of the oncologist and cardiologist. Clearly, the baseline measurement of LVEF before chemotherapy is most important in the evaluation of anthracycline-induced cardiotoxicity.
PATHOPHYSIOLOGY OF ANTHRACYCLINE-INDUCED CARDIOTOXICITY
The histological pathophysiology of anthracycline-induced cardiotoxicity is characterized by myocardial damage due to proteolysis, necrosis, apoptosis, and fibrosis. Proteolysis is a relatively acute response to anthracycline treatment. Treatment of cultured adult rat cardiomyocytes with doxorubicin for 24 hours resulted in the degradation of titin, the largest myofilament protein [15]. This is an early event after doxorubicin treatment, and proteolysis of the spring-like domain of titin may predispose cardiomyocytes to diastolic dysfunction and myofilament instability [15]. Electron microscopic examination of a doxorubicin-induced cardiotoxicity rat model demonstrated the destruction of the microstructures of myocardial cells, including myofilament dropout (Fig. 1A, upper arrow), loss of the Z-band (Fig. 1A, lower arrow), and vacuolation of mitochondria (Fig. 1B, arrow) [16]. These changes result in the dysfunction of myocardial cells and even cell death via necrosis.
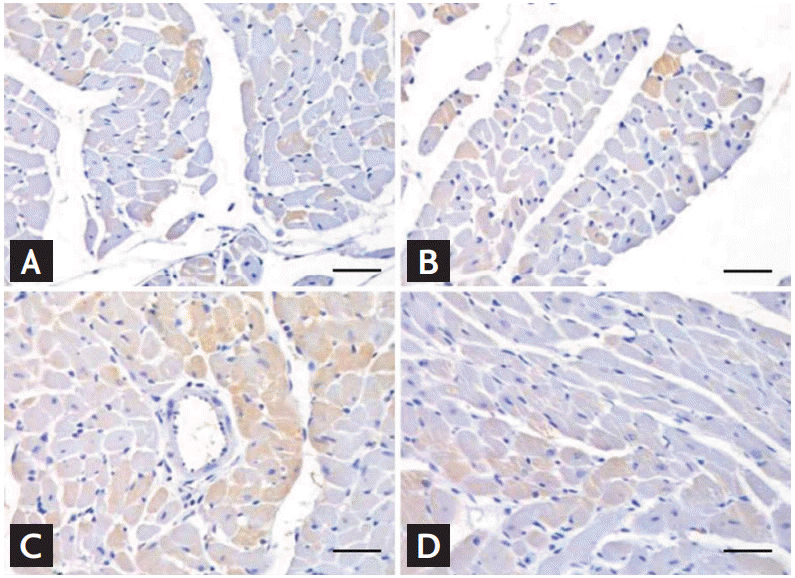
Apoptosis is another important mechanism involved in myocardial damage. After intraperitoneal injection of doxorubicin for 2 weeks, light microscopic examination of the rat myocardium revealed nuclear fragmentation and chromatin condensation by H&E staining (Fig. 2A) and Bax- (Fig. 2B), caspase 3- (Fig. 2C), and TUNEL (terminal deoxynucleotidyl transferase dUTP nick end labelin) assay- (Fig. 2D) positive apoptotic cells [17]. After the intraperitoneal injection of doxorubicin for 6 weeks in a chronic exposure animal model, the fibrosis burden was increased in the interstitial space of the myocardium of doxorubicin-treated normotensive Wistar-Kyoto rats (Fig. 3C) compared with control rats (Fig. 3A). The most extensive fibrotic burden was observed in the interstitial space of doxorubicin-treated spontaneously hypertensive rats (Fig. 3D) compared with the controls (Fig. 3B), suggesting that hypertension is a definite risk factor for doxorubicin-induced myocardial damage [18]. The same pattern of changes was also observed in the perivascular area. Early cell death and the delayed increment of myocardial fibrosis are major histological changes that eventually result in both systolic and diastolic cardiac dysfunction.
The molecular mechanism of cardiotoxicity has also been studied extensively. The most widely accepted mechanism is the generation of reactive oxygen species (ROS), related to oxidative stress [19]. During the metabolism of anthracyclines, unpaired electrons can be rapidly transferred to an oxygen molecule resulting in ROS generation [20]. The generation of superoxide anions by anthracycline metabolism can cause subsequent cellular damage by the degradation of the sarcomere, mitochondrial dysfunction, and DNA damage [21,22]. The reduction of the carbonyl group of anthracyclines generates toxic metabolites at the myocardium level [23]. The accumulation of toxic metabolites inhibits the calcium and sodium exchange pumps in the mitochondrial membrane, induces disturbance in the myocardial energetics and eventually systolic dysfunction [24]. Anthracyclines can also alter the iron homeostasis through the creation of Fe-anthracycline complexes, subsequently producing ROS [25]. Oxidative stress also mediates mitochondrial dysfunction by cardiolipin, a phospholipid with an important role in energy metabolism and a component of the inner membrane of mitochondria [26].
Anthracyclines can facilitate the release of pro-inflammatory cytokines by stimulating macrophages [27], which play a role in the development of cardiotoxicity. Interleukin (IL)-1╬▓ and IL-6 expression is increased in doxorubicin-treated cells, without changes in the expression of tumor necrosis factor (TNF). These cytokines mainly modulate apoptosis through TNF receptors, whose function is affected by doxorubicin [28]. According to our previous study, p53 could promote apoptosis by a caspase-independent pathway, and doxorubicin induced p53 expression in H9c2 cardiomyocytes [29]. Topoisomerase (Top) 2╬▓, a recently suggested primary mediator for anthracycline-induced cardiotoxicity, is required for p53 activation in response to anthracycline-induced DNA damage in cardiomyocytes [30]. Top 2╬▓ also reduced antioxidant enzyme gene transcription and induced ROS production [30] and deletion of the Top 2╬▓ gene protected the myocardium from anthracycline-induced cardiotoxicity in a mouse model [31].
Anthracycline-sensitive cardiac ankyrin repeat protein (CARP), a transcriptional regulatory protein that negatively regulates the expression of cardiac genes, is another molecule with a role in anthracycline-induced cardiotoxicity [32]. The expression of CARP in cardiac myocytes of spontaneously hypertensive rats was also reported (Fig. 4), suggesting that hypertension upregulates CARP expression. However, the role of CARP in the anthracycline-induced cardiotoxicity of hypertensive patients is unclear.
Damage to intracellular molecules by ROS and toxic metabolites of anthracyclines, the modulation of pro-inflammatory cytokines, and the interaction of anthracyclines with intracellular proteins such as Top 2╬▓ and CARP can lead to cardiomyocyte death. Because cell death plays an important role in the development of anthracycline-induced cardiotoxicity, the myocardial cell number determined during the embryonic development period can be an independent factor for the susceptibility of cardiac dysfunction [33]. Survivin, a key regulator of mitotic progression, plays a crucial role in controlling the cardiomyocyte number during embryonic development and adult life through its profound impact on cardiomyocyte replication and may thus emerge as a new target for myocardial regeneration [33].
STRATEGIES FOR CARDIAC FUNCTION MONITORING
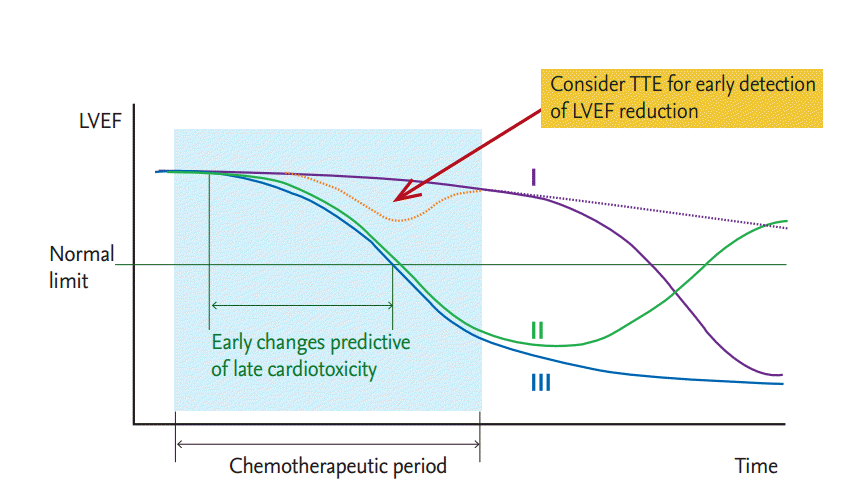
The cumulative dose of anthracycline is the most important risk factor for the development of cardiotoxicity. The estimated heart failure incidences were 5%, 16%, and 26% for cumulative doses of 400, 500, and 550 mg/m2 doxorubicin, respectively [34], and the limitation of the cumulative anthracycline dose is 400 to 450 mg/m2. The risk of cardiac events after chemotherapy is higher than tumor recurrence, with a 10-fold higher rate of cardiovascular disease and 15-fold increased rate of heart failure than in the general population [35]. Subclinical cardiotoxicity can develop during chemotherapy (Fig. 5) [7,36]; some patients may recover from it, while others may progress to overt heart failure [36].
Although it is not clear which clinical factor differs between the recovering group and progress-to-heart failure group, it is important to detect subclinical cardiotoxicity during the chemotherapeutic period. The cumulative incidences of cardiac events peaked at 1 year after the completion of anthracycline treatment [37,38]. There are insufficient data to determine the incidence of late-onset cardiotoxicity [2], which is defined as the development of cardiac events more than 1 year after the completion of anthracycline treatment. A significantly higher incidence of cardiovascular risk factors, such as hypertension, diabetes, dyslipidemia, and obesity in survivors of childhood cancer has been reported, and these factors may play a role in the development of late-onset cardiotoxicity [39]. Patients who already have cardiovascular risks before chemotherapy require more attention for cardiotoxicity in the course of treatment.
Because anthracycline-induced cardiotoxicity is suggested to cause irreversible damage to the myocardium (Fig. 5, line III) [36,40], the proper monitoring of cardiac function and early detection of cardiotoxicity are crucial. The assessment of baseline cardiac function before starting chemotherapy is the first step in monitoring. By definition, the detection of reduced LVEF compared with the baseline is a critical step in the diagnosis of cardiotoxicity. MUGA and echocardiography are the most studied methods for the measurement of LVEF. MUGA is somewhat insensitive for the detection of subtle changes in cardiac function [36]. Therefore, echocardiography is now the gold standard for screening and early detection of subclinical cardiotoxicity. Echocardiography has advantages in the assessment of diastolic dysfunction that can develop before systolic dysfunction; tissue velocity imaging and deformation imaging techniques, such as the strain and strain rate in both 2D and 3D echocardiography, can detect early subclinical changes in myocardial function [41,42].
Recently, cardiac magnetic resonance imaging (MRI) has been increasingly used for the evaluation of cardiotoxicity due to its accuracy and reproducibility in the measurement of the ventricular volume and LVEF [43]. MRI can also provide an opportunity for the detection of myocardial inflammation and edema during the early stages of cardiac injury and the presence of myocardial fibrosis in the late stages [44]. The limitation of using MRI for cardiac function monitoring is that it may not be cost effective for repeated assessments.
Circulating biomarkers, such as troponin and brain-type natriuretic peptide (BNP) are widely studied for use in the monitoring of cardiotoxicity. Troponin, the most widely studied biomarker for the detection of cardiotoxicity; it is recommended that troponin levels are evaluated at baseline and during and after chemotherapy [13]. The early elevation of troponin (i.e., within 72 hours after each cycle) and sustained elevation (i.e., 1 month after treatment) suggest the highest cardiotoxicity event rate [45]. However, the role of BNP in the prediction and diagnosis of cardiotoxicity remains unclear [46].
The 2012 European Society of Medical Oncology (ESMO) Clinical Practice Guidelines for cardiovascular toxicity [13] recommended management algorithms for anthracycline-induced cardiotoxicity. For cardiac function monitoring, it was recommended that the first follow-up echocardiogrphy should be performed at the end of chemotherapy and not at the cut-off cumulative dose of anthracycline. However, as the development of subclinical cardiotoxicity can occur at less than 300 mg/m2 of the cumulative dose of doxorubicin [7], cardiac function monitoring should be performed before the completion of anthracycline treatment. Although it was experience of only three hospitals in Korea, this study [7] demonstrated that, at the cumulative dose of doxorubicin of 244.5 mg/m2, the subclinical cardiotoxicity could be predicted (sensitivity, 71.4%; specificity, 70.9%; area under the curve, 0.741; 95% confidence interval, 0.608 to 0.874; p = 0.001) and suggested that performing early monitoring before reaching the 300 mg/m2 cumulative dose of doxorubicin might be a proper strategy for the early detection of anthracycline-induced cardiotoxicity. Additionally, in the same study, the enforcement rate of cardiac function monitoring with echocardiography was very low previously, only about 5 to 6 years ago; therefore, physicians should pay attention to cardiac function monitoring during the chemotherapeutic period. The guidelines also recommend an annual follow-up with echocardiography 1 year after the completion of anthracycline treatment but did not recommend how long cardiac function should be followed-up after chemotherapy.
A recent cardiac function monitoring study employed MRI and echocardiography 18 years after the completion of anthracycline treatment and demonstrated that the incidence of LVEF < 50% was 16% with both methods [47]. Considering that the patients received a relatively high dose of epirubicin, the incidence of cardiotoxicity was lower than early-onset cardiotoxicity within 1 year after completion of chemotherapy. This result indicates that some patients still need long-term monitoring of cardiac function, and further studies are mandatory to determine which patients and how long cardiac function should be monitored.
STRATEGIES FOR THE PROTECTION OF CARDIAC FUNCTION
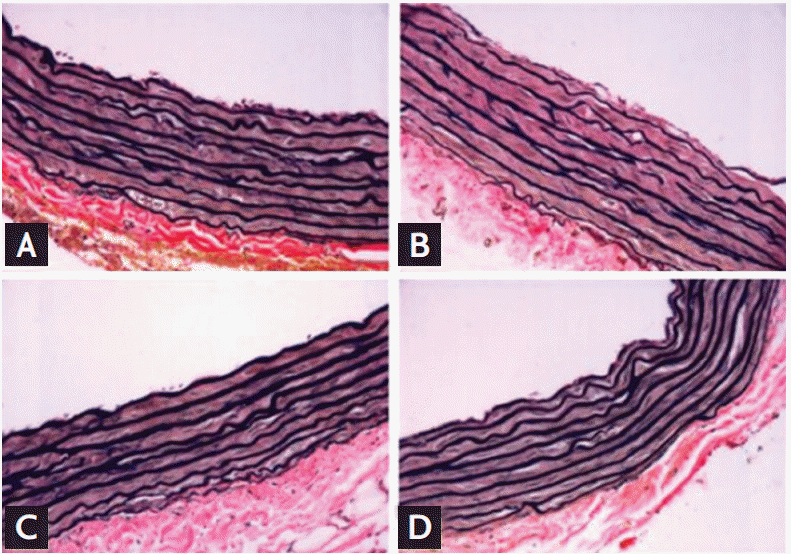
There are strategies for the protection of cardiac function during anthracycline treatment: primary and secondary prevention [4]. Primary strategies for the prevention of cardiotoxicity include the continuous infusion of anthracycline and the use of liposomal doxorubicin and the cardioprotective agent dexrazoxane. The continuous infusion of doxorubicin for 48 to 72 hours is effective for cardioprotection in sarcoma and lymphoma but not in pediatric patients. Liposomal doxorubicin is approved for limited cancer types. Dexrazoxane is approved only for women with metastatic breast cancer who received at least 300 mg/m2 doxorubicin and need additional doxorubicin for the maintenance of tumor control [4]. In animal studies, the concomitant administration of insulin-like growth factor 1 reduced apoptosis in doxorubicin-treated mouse myocardium [48], and candesartan prevented elastin degradation (Fig. 6) in doxorubicin-treated rat aorta [49]. In humans, many agents were studied for a possible cardioprotective effect against anthracycline-induced cardiotoxicity (Table 1) [50]; however, there are still limited data [48]. Secondary prevention strategies including the early detection and treatment of left ventricular dysfunction are suggested by the 2012 ESMO guidelines [13]. These guidelines recommended the aggressive treatment of left ventricular dysfunction even in asymptomatic patients after anthracycline therapy, particularly when the patient has a good chance for long-term survival.
Angiotensin-converting enzyme inhibitors, angiotensin receptor blockers, and ╬▓-blockers should be administered, and the earlier heart failure therapy is initiated (within 2 months from the end of anthracycline therapy), the better the therapeutic response will be [13]. Although starting treatment is recommended if left ventricular dysfunction is detected, many cancer survivors are not receiving treatment consistent with heart failure guidelines [11]. Oncologists should pay more attention to cardiac monitoring, and cardiologists should focus more on the proper treatment of anthracycline-induced cardiotoxicity.
CONCLUSIONS
Understanding the pathophysiology of anthracycline-induced cardiotoxicity is important to determine the therapeutic target of cardiotoxicity and for the development of protective agents. Drugs used in current clinical practice, such as dexrazoxane, ╬▓-blockers, angiotensin-converting enzyme inhibitors, and angiotensin receptor blockers, do not exert sufficient cardioprotective effects in anthracycline-induced cardiotoxicity; however, there are ongoing studies to evaluate proper treatment strategies. Cardiac function monitoring in the chemotherapeutic period is an important step for the early detection of anthracycline-induced cardiotoxicity, and treatment at the proper time can prevent progression to overt heart failure after anthracycline therapy to increase the odds of long-term survival. Collaboration between oncologists and cardiologists is necessary to improve the management of cancer patients receiving anthracyclines.



 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print





