


To the Editor,
The neurologic manifestations observed in allogeneic stem cell transplantation (allo-SCT) recipients are related to infections, metabolic abnormalities, and relapse of malignant disease in the central nervous system (CNS). After undergoing allo-SCT, graft-versus-host disease (GVHD) can become a serious problem that contributes to patient morbidity. GVHD is a systemic syndrome mediated by donor T cells and principally targets the skin, gut, and liver. Involvement of other organ systems has been described. Several reports have described that GVHD affects the CNS [1,2]; however, CNS-GVHD is a rare finding and remains controversial. Confounding factors, including infections, drugs, and autoimmune manifestations can make the diagnosis challenging. Among the reports, only a few small case reports have described histological findings. We experienced a case in which the symptoms mimicked those of CNS-GVHD after allo-SCT but were ultimately considered a newly developed case of multiple sclerosis (MS).
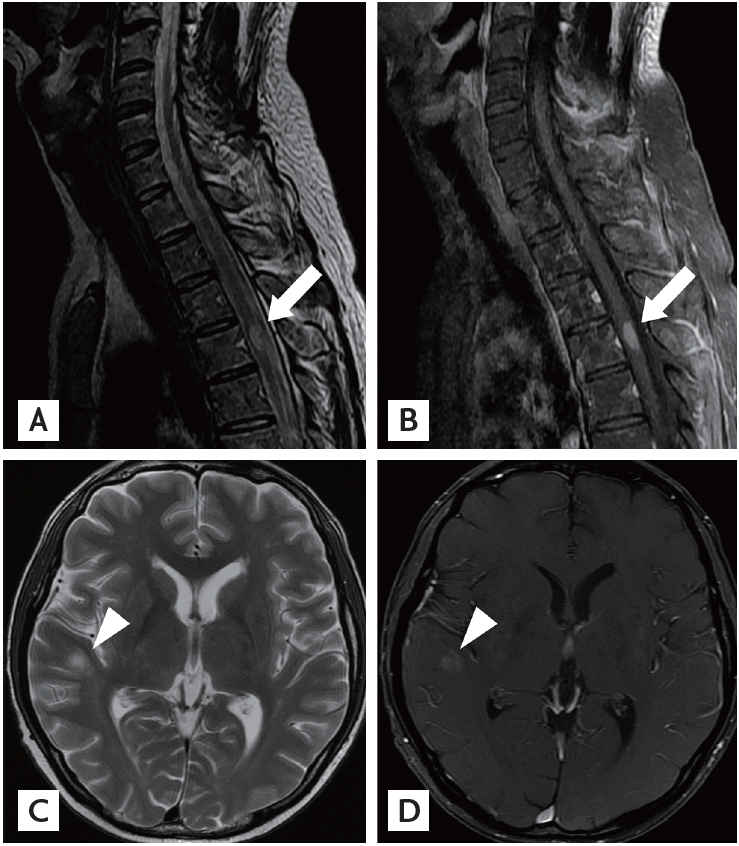
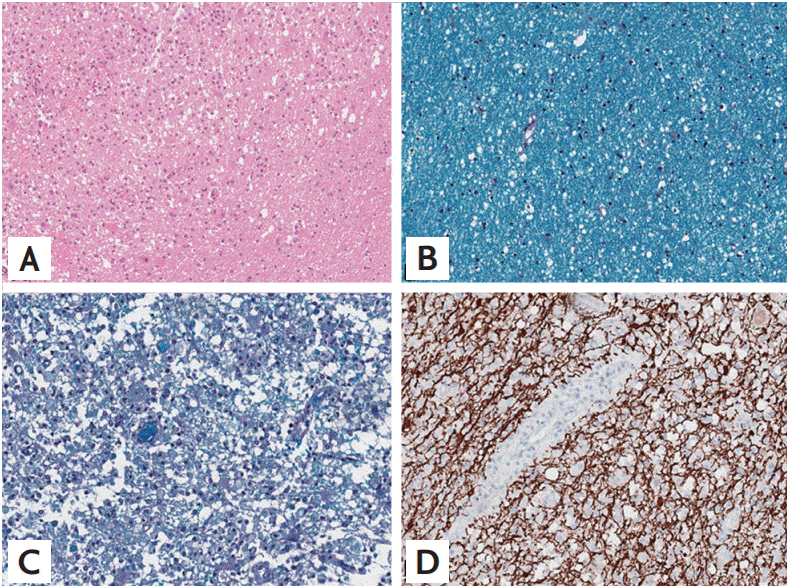
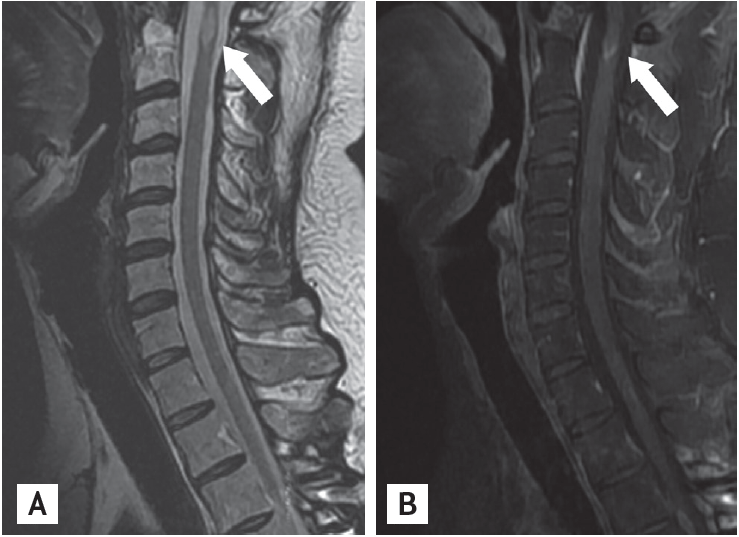
A 36-year-old man with no history of demyelinating disease or any other specific illness was diagnosed with acute lymphoblastic leukemia. The patient underwent matched related allo-SCT. Cyclosporin-A followed by tacrolimus were administered for GVHD prophylaxis. Approximately 8 months after allo-SCT, he was admitted to our hospital complaining of watery diarrhea. An endoscopic biopsy of the colon revealed multiple apoptotic bodies in the crypts and increased lymphoplasma cell infiltration in lamina propria, and he was diagnosed with gastrointestinal GVHD (GI-GVHD). High-dose oral steroids were administered and improved his symptoms. No eventful or GVHD-related disease was observed for 1 year after allo-SCT. Additionally, his bone marrow showed normal blood cells and complete donor chimerism. The patient presented 2 years after allo-SCT with a progressive tingling sensation below the abdomen and weakness in both legs. He also described progressive difficulty climbing stairs and walking. He did not complain of visual disturbances, dizziness, or headache. A neurological examination demonstrated symmetrical muscle weakness in both legs (Medical Research Council [MRC] grade 3) and increased deep tendon reflexes during induced knee and ankle reflexes. A sensory examination revealed decreases in all modalities below the T6 dermatome. Normal results were observed on the mini-mental state examination, as well as on cranial nerve and cerebellar function tests. No Babinski sign or ankle clonus were observed. Brain and spinal magnetic resonance imaging (MRI) revealed high T2 signal intensity lesions at T3 level of the spinal cord and of the corpus callosum and right temporal subcortex (Fig. 1). These lesions were enhanced with gadolinium. Cerebrospinal fluid (CSF) was clear and showed no pleocytosis, oligoclonal bands, or increased protein. Serum and CSF viral studies were negative for Epstein-Barr virus (EBV), cytomegalovirus, and the John Cunningham virus. There was no evidence of a CNS infection or leukemia relapse in the CNS. Furthermore, he had a GI-GVHD history; thus, we diagnosed probable CNS-GVHD. The patient received high dose intravenous methylprednisolone for 5 days followed by oral prednisolone (60 mg). Despite this treatment, his symptoms failed to improve. A stereotactic brain biopsy (right temporal lobe) revealed gitter cells (foamy macrophages) and loss of myelinated nerve fibers without leukemic infiltration (Fig. 2). An excision biopsy of the thoracic spinal cord revealed only fibrosis. His symptoms had improved gradually 3 weeks later. Strength in the lower extremities was MRC 4 and sensory numbness showed steady improvement. The patient was discharged and could ambulate independently. Oral prednisolone was tapered by 15 mg every other day over 8 months. The patient redeveloped a tingling sensation and weakness in all extremities approximately 3 years after allo-SCT. Neurological signs indicated weakness in bilateral arms (MRC grade 3) and legs (MRC grade 2) and decreased sensory responses below the T1 spinal cord level, and increased bilateral deep tendon reflexes in the upper and lower extremities. A spinal MRI revealed partially enhanced C1–2 lesions in the spinal cord (Fig. 3). Anti-nuclear antibody, anti-neutrophil cytoplasmic antibody, rheumatoid factor, anti-Ro, anti-La, cryoglobulin antibody, and anti-aquaporin-4 antibody were not detected. Both P100 latencies were delayed (right, 120 ms; left, 125 ms) in a visual evoked study. Furthermore, electrophysiologic evidence suggested central conduction delays in bilateral median and posterior tibial nerve evoked potentials. He was treated with high doses of intravenous methylprednisolone, and his symptoms improved gradually. Arm and leg weakness had become MRC 5 1 month later. We initially considered CNS-GVHD as a possible diagnosis based on the CNS characteristics. We could have also considered a CNS demyelinating disease, which can cause relapsing myelopathy, such as relapsing and remitting MS, as another possible diagnosis. The patient was started on subcutaneous injections of interferon β (IFN-β) in addition to oral prednisolone (10 mg). No eventful symptoms were detected that indicated a relapse during a 36-month observation period.
Although GVHD commonly affects the skin, gastrointestinal tract, liver, and hematopoietic system, some reports have described patients with CNS manifestations associated with GVHD and support the possibility that the brain is a target organ for GVHD based on animal studies [3]. However, among the reported antemortem diagnoses of possible CNS-GVHD, few had histological corroboration. This entity is particularly difficult to prove because differential diagnoses, such as CNS lymphoma, opportunistic infection, and EBV-related disorders, must be excluded. CNS-GVHD should be considered only if there is no evidence of other diseases with overlapping features, radiographic characteristics of CNS involvement, response to immunosuppressive therapy, and adequate documentation of a perivascular T-cell infiltrate after a histological and immunophenotypic evaluation.
The evaluation for infectious disease was negative in our patient. The histological results were not compatible with relapsed leukemia in the CNS. The patient developed GI-GVHD, as demonstrated by an endoscopic biopsy, and developed relapsing myelopathy and asymptomatic brain lesions. The initial diagnosis was CNS-GVHD; however, we considered MS as a probable diagnosis for several reasons. First, the immunohistochemistry of the brain biopsy was negative for infiltration of cytotoxic T-cells (CD+3) or apoptosis. Near-complete loss of myelinated nerve fibers was observed using Luxol fast blue stain, and axons were relatively spared. The pathological analysis of the brain biopsy was more compatible with a CNS demyelinating disease than with CNS-GVHD. Second, GI-GVHD affects only the CNS and does not involve other organs for approximately 5 years after onset. Third, MRI imaging revealed multiple white matter brain and spinal cord lesions. These separate episodes of neurological symptoms and radiological findings are characteristic of MS. Last, relapses continued to occur despite sufficient steroid treatment, whereas no relapses were observed for 36 months after the patient received IFN-β. IFN-β specifically targets MS and induces a more effective response against MS as compared to GVHD. Therefore, its application may be an important was to distinguish the two suspected diseases.
Autoimmune disorders may increase in incidence after allo-SCT. Myasthenia gravis, dermatomyositis, polymyositis, Guillain-Barre syndrome, and acute disseminated encephalomyelitis with an autoimmune component have been reported in patients after a bone marrow transplant (BMT). Demyelination disorders after BMT are very uncommon and only a few cases have been reported [4]. Histological findings suggestive of GVHD have been confirmed in a minority of reported cases, and the exact etiology of the demyelination remains largely undefined.
Immunotherapies, such as cyclophosphamide, mitoxantrone, chemotherapy, and allo-SCT have been evaluated to assess their efficacy for treating refractory MS, which affects only a small portion of patients with MS. Allo-SCT has been suggested as a potential treatment for MS and other autoimmune disorders that fail to respond to approved therapies. In contrast, our patient was a case of newly developed MS after allo-SCT. Because this was such a rare finding, we report it as an exemplary case. It is unfortunate that we have no information about whether the donor had a demyelinating disorder or developed MS after donating bone marrow.
One case study describing recurrent CNS manifestations after allo-SCT has been reported [5]. However, all cases were considered CNS-GVHD rather than MS based on clinical symptoms and histological findings. Distinguishing between CNS-GVHD and MS can be challenging, as the two diseases share similar clinical presentations and radiological findings. Furthermore, both CNS-GVHD and MS are rare in a clinical setting. However, the consequences of both diseases can be quite serious. Continued reporting and work-up of similar cases is required to develop strategies for a timely and accurate differential diagnosis.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print





