


To the Editor,
Wegener's granulomatosis (WG) is an inflammatory disorder of presumed autoimmune pathogenetic mechanism characterized by granulomatous inflammation of the respiratory tract, necrotizing vasculitis, and glomerulonephritis. However, the disease can affect any organ and shows diverse clinical manifestations. Although antineutrophil cytoplasmic antibodies (ANCA) are strongly associated with the pathogenesis and diagnosis of WG [1], diagnosis of the disease is difficult because it must be differentiated from an atypical infection or other autoimmune or hematologic diseases [2].
Here, we report a case of WG mimicking hemophagocytic lymphohistiocytosis, a clinicopathological entity involving the proliferation of hemophagocytic histiocytes in the bone marrow, resulting in fever, hepatosplenomegaly, lymphadenopathy, pancytopenia, liver dysfunction, and coagulopathy.
A 43-year-old female, previously diagnosed with hemophagocytic lymphohistiocytosis (HLH), was transferred from another hospital due to persistent fever. Her body temperature was 39.4℃ and had persisted for 5 days despite antibiotic treatment.
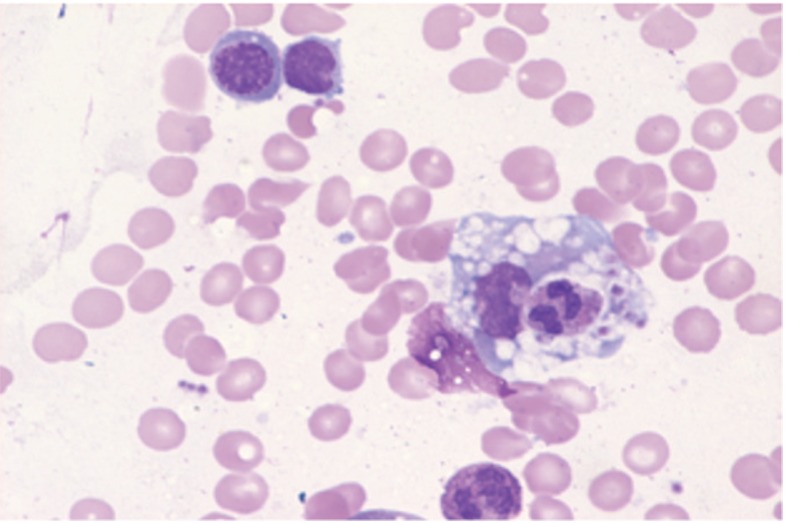
An inspection of the patient's clinical history revealed that in 2003 she had been diagnosed with thrombocytopenia, anemia, and splenomegaly, and a bone marrow biopsy showed histiocytic hyperplasia with hemophagocytosis (Fig. 1). Therefore, the patient was suspected of having HLH. However, she was discharged from the hospital after the fever and bicytopenia subsided following empirical antibiotic treatment. In 2009, the patient was again hospitalized at another institution because of fever. The fever persisted without evidence of infection, and hepatosplenomegaly was noted. Therefore, a differential diagnosis of fever and autoimmune disease was performed. However, testing for autoimmune antibodies, including fluorescent antinuclear antibodies and ANCA, was negative, and the patient underwent liver and bone marrow biopsies. The liver needle biopsy showed sinusoidal neutrophilic and histiocytic infiltration with microabscess formation, while the bone marrow biopsy showed dysplastic changes in the granulocytic lineage and histiocytosis of the bone marrow. Laboratory testing revealed the following: ferritin, 285 ng/mL; triglycerides, 285 mg/dL; and fibrinogen, 691 mg/dL.
Although natural killer (NK) cell activity and soluble CD25 were not examined, the patient's clinical features and laboratory findings, including the biopsy results, fit the 2004 HLH diagnostic criteria.
For the treatment of HLH, etoposide with steroids was applied. Despite treatment, the patient developed a fever. At that time, a pulmonary nodule and cervical lymphadenopathy were detected. For the evaluation of malignancy, the patient underwent a left supraclavicular node dissection and right middle lobe wedge resection at another hospital; however, the pathologic findings showed perifollicular histiocyte infiltration.
The patient's fever subsided the day following admission to our hospital. For the evaluation of recurrent histiocytosis, we assessed the patient's family history and performed an NK cell cytotoxicity test. However, the patient had no family history of a fever of unknown origin or HLH, and the level of NK cell cytotoxic activity was normal. A mutation analysis of the perforin gene was also negative. Although bicytopenia, splenomegaly, and a fever were noted, hyperferritinemia, hypertriglyceridemia, and hypofibrinogenemia were not found. Nevertheless, the patient complained of new symptoms, including red eye, a tingling sensation with mild numbness in both lower limbs, and mild hearing loss. For the red eye, the patient underwent an ophthalmologic consultation; her eye showed features of anterior uveitis, which required a differential diagnosis of vasculitic rheumatoid disease. An electromyogram showed lower lumbosacral radiculopathy, while audiometry revealed mild sensorineural hearing loss.
On computed tomography of the chest and abdomen, which was performed on admission, multiple small irregularly shaped nodules were detected in both lungs, combined with multifocal intralobular ground glass opacities and mild interlobular septal thickenings. In addition, hepatomegaly was noted with decreased parenchymal enhancement in both kidneys.
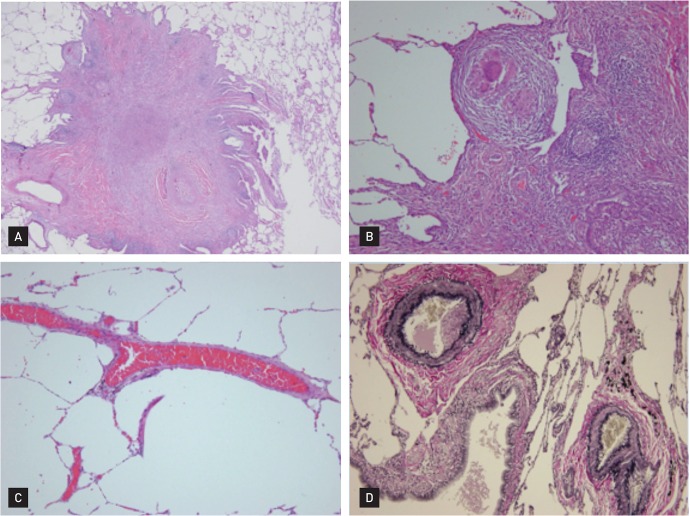
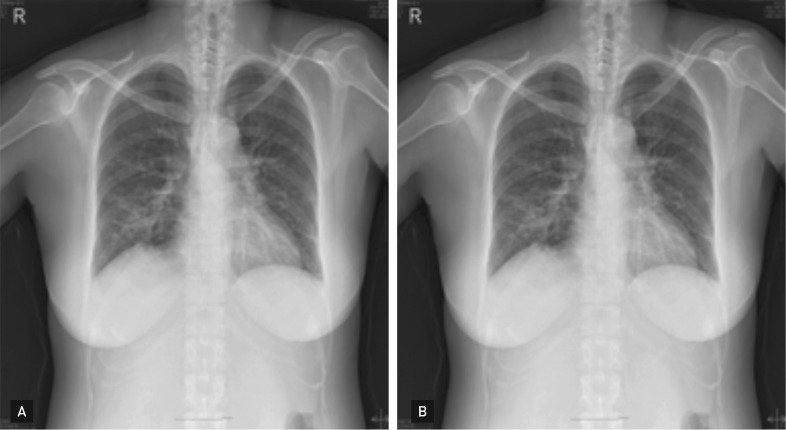
Given this clinical history, a test for autoantibodies was requested; the results revealed ANCA with a cytoplasmic pattern (c-ANCA), and an antiproteinase 3 antibody titer of 14.1 U/mL (negative, < 5). The pathology of the right middle-lobe wedge resection, which had been performed at another hospital, was reviewed, and a basophilic microabscess, granuloma, and vasculitis involving arteries and veins, dense fibrosis, and fibrinous vascular thrombosis were detected (Fig. 2). These results, coupled with anatomic pathological studies, suggested WG. As a preliminary treatment, a dose of 1 mg/kg/day of prednisolone was prescribed, and rapid clinical improvement in the patient's general health was noted with progressive recovery of her pain and hearing. After 3 months of treatment with prednisolone, the patient's general health improved; this was confirmed by chest radiography (Fig. 3).
WG is an uncommon autoimmune disease characterized by granulomatous inflammation of the respiratory tract and systemic small vessel vasculitis that is usually associated with significant morbidity and mortality. Although WG is considered to be an autoimmune disease, its pathogenesis is not fully understood.
The diagnosis of WG is made based on clinical features of illness associated with the presence of c-ANCA and vasculitis in small arteries and veins, with the presence of giant cells and epithelioid cell granulomas.
Although c-ANCA is not a diagnostic criterion of WG according to The American College of Rheumatology, it has 96% specificity and 92% sensitivity in the diagnosis of WG and is known to play a crucial role in the pathogenesis of WG. Nonetheless, if testing for c-ANCA is negative, it does not necessarily exclude disease; in fact, in the early stages of WG, testing for c-ANCA may be negative, especially if there is no renal involvement. In any case, c-ANCA is an important sign for the diagnosis and monitoring of the regression of the disease once treatment has been initiated.
Symptoms such as recurrent inflammation of the upper respiratory tract, otitis media, sinusitis, epistaxis, and saddle nose are critical information in the initial phase of WG. Further, if the disease progresses to generalized WG, fever, malaise, weight loss, and progressive glomerulonephritis can occur [3]. However, if these typical symptoms and serologic markers do not appear, the diagnosis of WG can be difficult and delayed. Our patient initially presented with a febrile episode without evidence of infection. Testing for autoimmune markers, including c-ANCA, on admission was negative. The only noticeable findings were bicytopenia and bone marrow histiocytic hyperplasia with hepatosplenomegaly, which could suggest hematologic disease, including HLH.
To date, hematologic and bone marrow findings of WG have not been studied and cytopenia and HLH cases associated with WG have been reported infrequently across the globe [4].
HLH is a potentially life-threatening hyper-inflammatory syndrome characterized by histiocyte and macrophage activation, and hemophagocytosis in the bone marrow and other reticuloendothelial systems. Although the precise pathogenetic mechanism of HLH is unknown, massive cytokine release from activated lymphocytes and macrophages and consequent histiocytic hyperplasia with hemophagocytosis are considered the main etiology.
HLH is not a single disease but a spectrum of closely related disorders having the features described above with two primary forms according to the etiologic factor: primary/familial HLH and acquired/secondary HLH.
Familial HLH is caused by a genetic aberrancy, such as a mutation in the perforin gene, resulting in reduced NK cell cytotoxicity. It mainly affects infants with recurring episodes and a fatal outcome [5]; however, secondary HLH is caused by an overreactive inflammatory process beyond regulatory capacity, which is initiated by various primary inflammatory triggers and which usually resolves after the primary causes are eliminated.
According to the causative factor, secondary HLH can be classified as virus-associated HLH, other infection-associated HLH, or malignancy-associated HLH [5]. Autoimmune diseases are included among these triggering factors; in such cases, the term macrophage activation syndrome is applied.
Even in secondary HLH, reduced NK cell cytotoxic activity and the proliferation of Th1-deviated immune responses are presumed to be a pivotal etiology. When the effective elimination of intracellular organisms is impossible or when antigenic stimulation causes continuous immune system activation, uncontrolled cytokine release provokes histiocytic proliferation and differentiation.
The pathogenesis of WG is similar to that of HLH in that both are caused by the hyperactivation of Th1-deviated immune responses and they both show histiocytic hyperplasia. However, overt HLH combined with WG is rarely reported compared to other autoimmune diseases, such as systemic lupus erythematosus and rheumatoid arthritis.
The reason is thought to be related to the mechanism of ANCA-associated vasculitis, which preferentially involves vessels of a specific size or specific organs. In the pathogenesis of WG, ANCA play a key role by promoting interactions between neutrophils and integrin proteins on vascular endothelial cells. Therefore, vascular endothelial cells are an important part of the pathogenesis of WG, and heterogeneity among the endothelial cells in different organs may be the basis of organ tropism in WG [3].
Nonetheless, WG is also a systemic disease that can lead to systemic, cytokine-mediated reactions. Because bone marrow contains a range of blood vessel types, including small arteries, it could be susceptible to the same diseases that affect blood vessels elsewhere in the body. Therefore, we assume that hematologic reactions mimicking or triggering HLH are possible. However, finding evidence of vasculitis or a granuloma in bone marrow by a blind biopsy in WG patients is thought to be difficult. The present patient presented with histiocytic hyperplasia on repeated bone marrow biopsies without vasculitis or granulomas. Moreover, she did not initially complain of any classic symptoms of WG. This is probably associated with the initial negativity for ANCA. In 2009, the reported clinical findings and laboratory results for the patient met the five provisions outlined in the 2004 HLH diagnostic criteria. However, because ferritin and triglycerides are easily affected by other conditions and given that the severity of the episodes was not intensive, the diagnosis of HLH is doubtful.
From our analysis of NK cell cytotoxic activity and the levels of ferritin, triglycerides, and fibrinogen, which were within normal limits, reactive bone marrow changes due to WG seem to be more reasonable.
Despite specific serologic markers, WG can be difficult to differentiate from certain systemic diseases. To properly diagnose WG, one must have a high degree of suspicion, especially in patients with refractory disease, with the involvement of numerous organs and systems and a worsening of the patient's general status.
Furthermore, although hematologic manifestations are uncommon, one should be aware that histiocytic hyperplasia in the bone marrow mimicking HLH can be a clinical manifestation of WG.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print





