


INTRODUCTION
A number of case reports have examined the association of moyamoya disease with systemic lupus erythematosus (SLE)1–3). Among these, one was a case of SLE and Sjögren syndrome associated with unilateral moyamoya disease1), and another was a case of intraventricular hemorrhage and cerebral ischemic attacks in the presence of lupus anticoagulant mimicking moyamoya disease2). Yet another study reported a case of SLE associated with bilateral moyamoya disease3). Moyamoya disease is a rare, progressive cerebrovascular disorder characterized by stenosis or occlusion of the terminal portions of the bilateral internal carotid arteries and abnormal vascular network in the vicinity of the arterial occlusion4). The common symptoms of moyamoya disease in children are stroke or recurrent transient ischemic attacks, such as disturbed consciousness and seizures, while symptoms in adults are intracerebral or subarachnoid hemorrhage4, 5).
Here we report a case of SLE with familial moyamoya disease in a young female patient who developed a severe migraines type headache which was not typical of moyamoya disease.
CASE REPORT
A 13-year-old girl had been well until February 15 2000, when she developed severe headaches and began vomiting. The severity of her headaches increased over two weeks, prior to admission. In March 2000, she was admitted to our hospital with intractable headaches. At age 12, she was diagnosed with SLE based on the findings of thrombocytopenia (51,000/μL), positive reaction for antinuclear antibody (1:640, speckled pattern), recurrent oral ulcer, proteinuria on urinary dipstick test (3+) and arthritis according to the ACR criteria6). Her past medical history was unremarkable. However, within her family, her mother was diagnosed with moyamoya disease several years ago for which she was treated with antimalarial drugs and a low dose of steroids until she suffered headaches because of the absence of SLE activity.
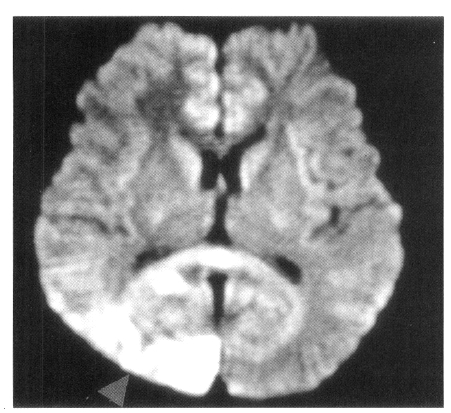
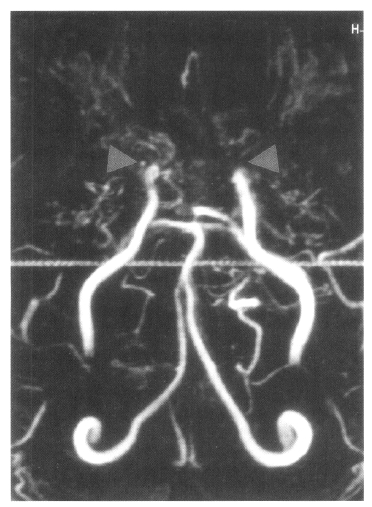
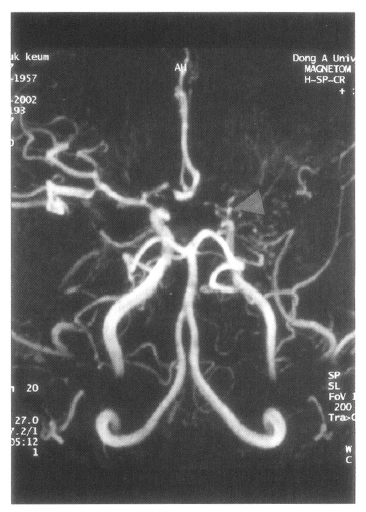
On admission, physical examination of the patient yielded the following data: temperature of 36.4°C, blood pressure 110/70 mmHg, pulse rate 78/min, and respiration rate 20/min. Physical and neurological examinations were unremarkable. The laboratory data revealed a white blood cell count 7,160/μL, hemoglobin 14.2 g/dL, reticulocytosis 1.1%, and platelet count 154,000/μL. Erythrocyte sedimentation rate (ESR) was 2 mm/hr. Chemical analyses of serum were within normal limits. C-reactive protein was 3.2 mg/L (normal <5), and a test for the veneral disease research laboratory (VDRL) turned out negative. Antinuclear antibody titer was 1:640 (speckled pattern) and anti-ds DNA antibody was 14.8 IU/mL (normal, <25 IU/mL). Anti-Ro, anti-La, anti-Sm, and anti-RNP antibodies were all negative. Complement levels were normal: C3 92.1 mg/dL (normal, 90–180), C4 12.6 mg/dL (normal, 10–40), and CH50 41.2 U/mL (normal, 30–45). Tests for anticardiolipin antibodies (IgG, IgM) and lupus anticoagulants were negative. A lumbar puncture also showed normal findings. Cerebrospinal fluid (CSF) cultures for bacteria and mycobacteria were all negative. We found a low-density lesion on brain computed tomography (CT) in the right temporo-occipital area, which indicated right posterior cerebral artery infarction. Magnetic resonance imaging (MRI) showed a high signal intensity lesion on the right posterior cerebral artery territory (Figure 1), and magnetic resonance angiography (MRA) revealed the occlusion of both the internal carotid artery without any visualized anterior cerebral artery, main trunk of middle cerebral artery and basal moyamoya vessels on both sides (Figure 2). Also, there was no stenosis of the internal carotid artery bifurcation with occlusion of anterior cerebral artery and main trunk of middle cerebral artery in the left side on her mother’s MRA (Figure 3). Cerebral angiography was not performed due to the patient’s refusal.
Since there was no evidence of disease activity or antiphospholipid syndrome, she was given conservative treatment and supportive medication, antidepressants, muscle relaxant, analgesics and low doses of aspirin. One week after administering the drugs, the patient had fewer headache. Furthermore, the patient suffered only mild headaches during 2-years of follow-up.
DISCUSSION
SLE is a disease characterized by a broad clinical spectrum and an array of immunological abnormalities. Involvement of the nervous system is reported to occur in up to 50% of patients preferentially as manifestations of the central nervous system often termed neuropsychiatric SLE (NPSLE)7, 8). Headaches are considered important symptoms in patients with SLE, occurring in a significant proportion of patients9). Some speculate the existence of a genuine lupus headache, severe, disabling, persistent, and headache not responsive to narcotic analgesics10). This form of headache has even been included in the criteria for assessing disease activity of SLE11). Whether headaches are a unique symptom attributable to SLE is not clear, but various underlying conditions, such as lupus cerebritis, transverse myelitis, pseudotumor cerebri, cerebral vasculitis, intracranial aneurysm and meningoencephalitis may be the cause of headaches.
The seriousness of the patient’s headaches initially suggested a “lupus headache”, although studies indicate that it is not specifically related to SLE expression or severity7, 9). Brain CTs showed a low density lesion in the right temporo-occipital area, which indicated infarction of the right posterior cerebral artery, but there was no evidence of disease activity and other causes of occlusion of the cerebral vessels. MRIs and MRAs showed typical bilateral moyamoya disease.
The etiology of moyamoya disease is still unclear, but frequent familial incidence suggests that some genetic factors may play a part. It occurs more frequently in females, and peak incidences are among persons aged 1–10 and 30–40 years. Approximately 10% of the cases in Japan have a family history of the disease. Analysis of the human leukocyte antigen (HLA) class II gene in Japanese patients with primary moyamoya disease revealed increased frequencies of DPB1*1601 and DBP1*0901 in early-onset (age<=10) patients and DRB1*0201 in late-onset (age>11) patients12). Han et al. reported familial incidence to be 1.8% in Korea, with age distribution showing two peaks. The patients under 10 years of age made up the first peak and patients from 20 to 39 years old made up the second peak13). Our patient and her mother had moyamoya disease. Her mother had a history of cerebral hemorrhage several years ago. HLA typing was not performed.
Although surgical management aims to enhance cerebral perfusion, medical management of moyamoya disease includes vasodilators, corticosteroids, or agents that reduce viscosity of blood cell agglutination. With the use of conservative measures, children may still have persistent cerebrovascular insults. Although children with moyamoya disease have a future risk for intracranial bleeding on lifelong anticoagulant therapy, even 10-year follow-up studies have not determined whether surgical revascularization would decrease the risk of recurrent bleeding after an episode of intracerebral hemorrhage14). We gave our patient conservative treatment and she responded to medical therapy. We did not use a vasodilator due to the risk of exacerbating the headaches.
In conclusion, we have described a case of SLE in a young female patient with a family history of moyamoya disease, who developed severe migraine-type headaches. We also mentioned the significance of moyamoya disease association with SLE. We suggest that moyamoya disease in association with SLE should be considered in the differential diagnosis of cerebrovascular events in patients with SLE.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print





