


INTRODUCTION
Kimura's disease (KD) is a rare benign disorder of unknown etiology, and is observed primarily in young Asian men. KD may the result of an allergic or chronic inflammatory response, preferentially occurring in the head and neck. The clinical symptoms are usually manifest either as painless unilateral lymphadenopathy, predominantly in the head or neck region, or elevated serum immunoglobulin (Ig) E and eosinophilia1,2). KD may also be associated with renal disease. Nephrotic syndrome is the most common manifestation of renal disease. KD lesions usually precede or coincide with the development of renal disease2,8).
Early diagnosis and recognition of KD may spare both patient and doctor from the need for unnecessary invasive diagnostic procedures. In the present case, the patient exhibited an unusual case of KD, occurring during hemodialysis, involving the thoracic and abdominal lymph nodes, which resolved itself spontaneously, and nearly completely, after two months.
CASE REPORT
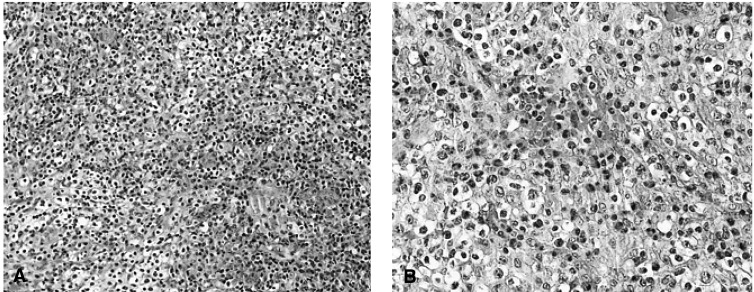
A 45-year-old man was admitted for evaluation of a series of painless, firm, movable neck masses. He had a history of tuberculous pleurisy 25 years prior to this complaint. He was also a carrier of hepatitis B virus surface antigen. He denied experiencing asthma, urticaria, or any other allergic reactions. He had received maintenance hemodialysis for 3 years, for end-stage renal disease resulting from chronic glomerulonephritis. Hemodialysis was performed 3 times weekly for 4 hours per session, using polysulfone dialysis columns. In the preceding 4 months, the patient had discovered masses in his neck. These masses had gradually increased in size. The patient had no history of fever, cold sweating, or weight loss. During the physical examination, firm and movable masses, measuring up to 3├Ś3├Ś2 cm (the largest one) were found. These masses were palpable with no tenderness, and were located primarily in the right anterior triangle of the neck. Although there were also several palpable masses in both the axillary and inguinal areas, no hepatosplenomegaly was detected. No warm sensations or erythema manifested on the overlying skin. Stool examinations, including ova and parasite evaluations, were negative. Laboratory findings included a hemoglobin level of 7.3 g/dL, platelet count of 200├Ś109/L, and a white blood cell (WBC) count of 6.2 ├Ś 109/L: the differential WBC count revealed 68.9% segmented neutrophils, 10.2% lymphocytes, 8.4% monocytes, 12.1% eosinophils, and 0.4% basophils. Serum total IgE level was 261.0 IU/mL. The blood urea nitrogen and serum creatinine level were 56 mg/dL and 4.8 mg/dL. Neoplastic disease appeared to be the most likely differential diagnosis. Fine needle aspiration cytology of the neck mass revealed findings of nonspecific reactive lymphadenitis. A computed tomography (CT) scan of the neck and chest revealed masses adjacent to both sternocleidomastoid muscles, numerous lymph nodes of varying size in both axillary areas, and over lymph nodes of over 1 cm in size in the paratracheal, subcarinal, and intrapulmonary areas (Figure 1A). A CT scan of the abdomen and pelvis displayed multiple lymph nodes of varying size, which were enhanced in the hepatic hilum, peripancreatic area, celiac axis, and paraaortic area (the largest was 4.5 cm) (Figure 1B). A tumor scan using Gallium-67 demonstrated more noticeable increased uptake lesions in the left lower cervical, left axillary, paraaortic, right iliac, and both inguinal lymph nodes (Figure 2). The findings from fine needle aspiration cytology were inconclusive with regard to diagnosis, thus an open biopsy was recommended. An excisional biopsy of neck masses and inguinal lymph nodes was performed. Results of histopathological examinations of the neck mass and lymph nodes revealed dense infiltration of eosinophils, lymphocytes, and histiocytes, along with many small vessels, mild fibrosis, and results consistent with KD, with no evidence of malignant lymphoma (Figure 3).
Thereafter, the patient has received no specific treatment, as his symptoms, with the exception of the mass, were clinically unremarkable. After two months, physical examination revealed no neck masses, and a follow-up abdominal CT scan showed that the numerous lymph nodes of varying size had subsided spontaneously (Figure 4). Until now, the patient has been progressing well on maintenance hemodialysis.
DISCUSSION
KD is a rare, benign condition, usually observed in young Asian men. Although its etiology remains unknown, it has been tentatively suggested that KD is the result of an aberrant allergic or chronic inflammatory response to viral, bacterial or parasitic infection3-5). KD usually manifests as a tumor-like lesion, showing a predilection for head and neck sites. Systemic manifestations of KD are characterized by elevated serum IgE, eosinophilia, and renal diseases, including focal segmental glomerulosclerosis, IgA nephropathy, minimal change disease, membranous nephropathy, and IgM nephropathy6-8). Clinical symptoms of KD normally precede or coincide with the development of renal disease2,8). Neck masses that were incidentally detected in end-stage renal disease patients were first considered in terms of differential diagnoses, possibly as neoplastic disease or infection, such as tuberculosis, in Asian patients. Clinically, KD may often be confused with malignant lymphoma or other metastatic cancers9). Consequently, a patient with KD is often unnecessarily evaluated for symptoms of far more serious diseases10). It is difficult to diagnose KD without a tissue biopsy, and fine needle aspiration cytology has only limited value in such cases. In the present case, neoplastic disease was considered before any other diagnosis. During the CT scan, we were also unable to differentiate these lesions from malignant lymphoma or metastasis. Unlike most cases, our patient exhibited multiple lymphadenopathy, including thoracic, abdominal, neck, axillary, and inguinal lymph nodes. KD was confirmed by excisional biopsy on three sites in our case. Histopathological analyses of the neck mass and lymph nodes revealed no signs of malignant lymphoma. Clinicopathologically, KD must be distinguished from angiolymphoid hyperplasia with eosinophilia (ALHE). KD occurs predominantly in Asians, and preferentially in males. Patients invariably exhibit peripheral eosinophilia and elevated serum IgE levels. In contrast, ALHE occurs in all racial groups, showing a slight predominance in females11). Regional lymphadenopathy, serum eosinophilia, and elevated IgE levels are rare in cases of ALHE. Both diseases are characterized by increased eosinophilic infiltration and vascular proliferation. KD typically manifests several histologic features, such as predominant eosinophilic infiltration with follicular hyperplasia, fibrocollagenous change, and vascular proliferation. However, vascular proliferation is more significant in ALHE. In ALHE, vessels are aggregated and lined by plump cuboidal, or occasional hobnail, endothelial cells, with frequent cytologic atypia and vacuolization. However, these angiomatoid features are never observed in cases of KD12). In our case, we observed no vessels lined by plump cuboidal endothelial cells suggesting ALHE, and there were characteristic and distinctive clinical features suggesting KD. Therefore, we excluded ALHE as a diagnosis.
Renal manifestations in KD include visceral localization. We have no evidence that would explain this relationship between KD and end-stage renal disease in our patient. Our patient represents a very unusual case of KD, occurring during hemodialysis. The treatment of KD involves one of three major approaches: surgical excision, irradiation, or steroid therapy. The clinical course of KD is generally benign. Our patient had no complaints or symptoms, with the exception of the aforementioned palpable masses. Thus, the patient has received conservative treatment while undergoing regular checkups.
In conclusion, a high index of suspicion regarding KD is highly recommended. The early diagnosis and recognition of KD may spare both patient and doctor from the need for unnecessary invasive diagnostic procedures. To our knowledge, this is the first case of KD occurring during hemodialysis, involving thoracic and abdominal lymph nodes, which resolved itself both spontaneously, and nearly completely, after two months.



 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print





