


INTRODUCTION
Since D-penicillamine was introduced for the treatment of WilsonŌĆÖs disease, cystinuria, and rheumatoid arthritis, there have been reports of side effects which have limited its usefulness1). Many autoimmune syndromes have been associated with its use, including GoodpastureŌĆÖs syndrome, pemphigus, lupus erythematosus, Sj├ČgrenŌĆÖs syndrome, myasthenia gravis2), and polymyositis/dermatomyositis3ŌĆō5).
We encountered a case of polymyositis in a patient with primary biliary cirrhosis who had been treated with D-penicillamine. Polymyositis improved promptly after prednisolone therapy along with discontinuation of D-penicillamine.
CASE REPORT
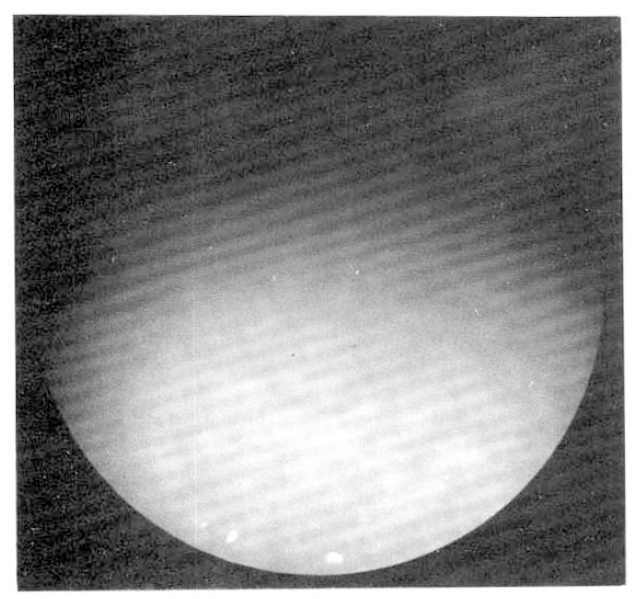
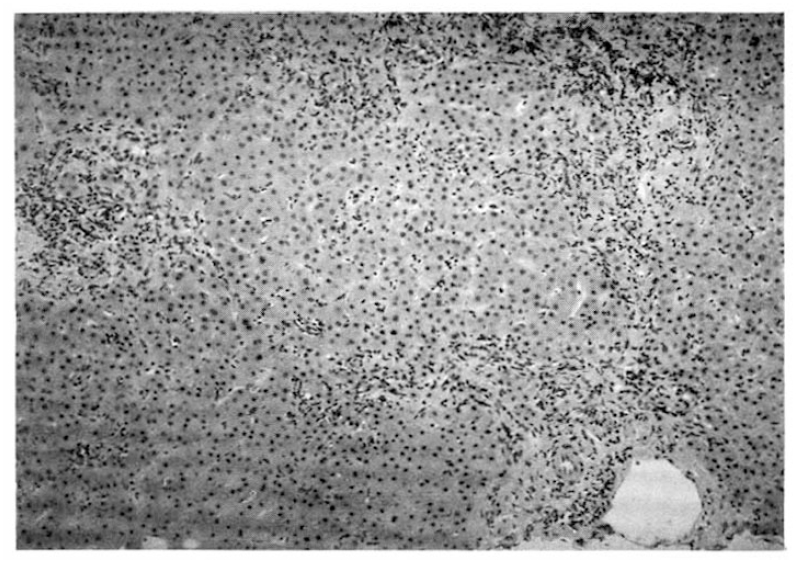
A 55-year-old woman was admitted to Seoul National University Hospital, because of general weakness and abdominal distension in June, 1992. The patient was relatively healthy until 1983 when she developed generalized pruritus and fatigue. In 1987, she was admitted to the Seoul National University Hospital and workup included serologic markers for Hepatitis B, which were negative, gastrofiberscopy, peritoneoscopy (Fig. 1), liver biopsy (Fig. 2), and antimitochondrial antibody (1:1280 positive) to be diagnosed as primary biliary cirrhosis. She was treated with D-penicillamine at the dose of 750 mg per day. This regimen continued for 1 year without apparent complication until she found generalized edema with proteinuria (3808 mg/day). D-penicillamine was discontinued for 1 month. When the urine became normal, D-penicillamine was reintroduced at the same dose without problem. In March 1992, she noted general weakness and flu-like illness. Her weakness had been progressively aggravated and 1 week before admission she noticed abdominal distension with febrile discomfort.
Upon admission, she complained of generalized weakness, abdominal discomfort and also decreased muscular power of the extremities. She had difficulty in getting out of bed, walking and climbing stairs. She was unable to sit up without assistance.
On examination, the temperature was 37.5┬░C, the pulse rate 80 per minute and the blood pressure 100/60 mmHg. She appeared chronically ill, and the skin was dry and dark. The head and neck was normal without lymphadenopathy or thyromegaly. The lungs were clear and the heart was normal. The abdomen was mildly distended with shifting dullness, but not tender. The extremity showed symmetrical proximal muscle weakness (Grade III/V) in the neck flexor, triceps, biceps, hip flexor, knee extensor and flexor. Sensory examination was grossly intact and deep tendon reflex was normal.
The Westergren erythrocyte sedimentation rate was 65 mm/hour, hemoglobin 9.8 g/dl; hematocrit 29.6% (MCV 95.3 fl, MCH 32.3 pg, MCHC 33.9 g/dl, RDW 15.0%); white cell count 7300/mm3 (neutrophils 80%, lymphocytes 16%, monocytes 4%); platelet count 120,000/mm3. The urine was normal. Serum cholesterol was 173 mg/dl; total protein 5.7 g/dl; albumin 2.1 g/dl; bilirubin 1.0 mg/dl; alkaline phosphatase 907 U/L (normal 30 to 115); AST 682 U/L (normal less than 40); ALT 333 U/L (normal less than 40). Serum creatine kinase was 6,400 IU/L (normal 20 to 270), lactate dehydrogenase 1480 IU/L (normal 10 to 225). Antinuclear antibody was positive at 1 : 1280 dilution (nucleolar pattern). Serologic markers for Hepatitis B or C were negative.
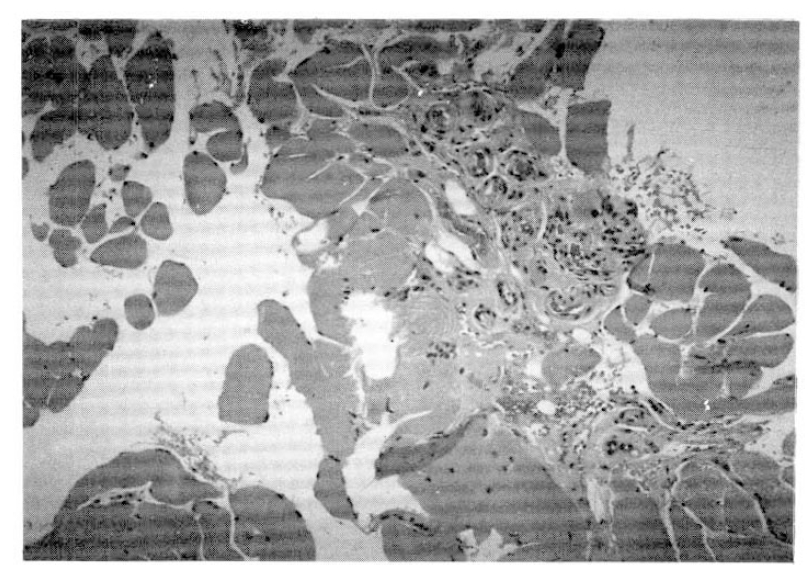
The electromyography revealed increased insertional activity, fibrillation, and complex repetative discharge of small amplitude and polyphasic motor unit potentials, compatible with inflammatory myopathy. The muscle biopsy revealed multifocal degenerating and regenerating myofibers with infiltration of inflammatory cells (Fig. 3).
She was diagnosed as polymyositis and liver cirrhosis with portal hypertension, and evaluated looking for underlying malignancy. Workup included mammography, gastrofiberscopy, stool exam, abdominal ultrasonography, which were all negative. She was managed with diuretics for the control of ascites and corticosteroid therapy (prednisolone 50 mg per day) along with the discontinuation of D-penicillamine. The level of creatine kinase, lactate dehydronase, AST, and ALT were decreased to 56 IU/L, 298 IU/L, 67 U/L, and 16 U/L respectively in 3 months with clinical improvement.
DISCUSSION
D-penicillamine has side effects including dermatitis (12ŌĆō25%); anorexia, nausea, vomiting and diarrhea in 12ŌĆō20%; thrombocytopenia in 5ŌĆō10%; proteinuria in 10ŌĆō20%; myelotoxicity6). Autoimmune syndromes develop rarely during D-penicillamine therapy and improve after the drug is stopped; example include myasthenia gravis, polymyositis, GoodpastureŌĆÖs syndrome and systemic lupus erythematosus2,7,8). The frequency of polymyositis/dermatomyositis has varied between 0.2ŌĆō1.1% in previous reports9,10).
The development of polymyositis associated with D-penicillamine appears to be an idiosyncratic reaction that dose not have any relationship to dose or duration of administration. Some patients reported to develop polymyositis after 4 weeks or less, whereas other patients became symptomatic after 5 years or more of D-penicillamine therapy9,11,12). Kay (1986)13) reported that in patients with polymyositis/myasthenia gravis, the mean dose of D-penicillamine was 500 mg per day (range; 75 to 750 mg); mean duration of treatment 11.5 months (range; 2 to 27 months). In our patient, the duration and the cumulative dose of therapy prior to the diagnosis of polymyositis were 5 years and 1,100 g.
Although the exact etiopathogenesis of D-penicillamine-induced polymyositis is not known, it is thought that some effects of D-penicillamine on the humoral immunological system might play some role4). According to the studies, which reported that the toxic reaction of penicillamine was related to increased frequency of DRw2, DR3, and B8, it may be that some patients are genetically predisposed to develop D-penicillamine-induced polymyositis10,14ŌĆō17).
The D-penicillamine has two effects on humoral immunologic system; D-penicillamine dissociates IgM antibodies into inactive monomers, but at the same time markedly enhances the affinity of IgG to double-stranded DNA and induces the formation of small stable immune complexes; destruction of rheumatoid factor by D-penicillamine may produce direct antigenicity of these disorders4).
The clinical, laboratory and pathological features of our patient were indistinguishable from those seen in idiopathic polymyositis12,18). One study suggests that D-penicillamine-induced polymyositis show more rapid recovery after discontinuation of D-penicillamine and more benign course10).
Doyle et al (1983)12) reviewed 14 cases of polymyositis/dermatomyositis during the treatment of D-penicillamine. 12 patients recovered after D-penicillamine was withdrawn: 2 patients died from cardiac involvement. Since D-penicillamine-induced polymyositis can emerge at any time during the course of therapy and with any dose, it is prudent to monitor patients receiving D-penicillamine closely. Should polymyositis develop, it is generally advisable to discontinue the treatment. In view of the risk of singificant morbidity and mortality, specific treatment shoud not be delayed18). Rechallenge with D-penicillamine is generally discouraged as there have been reports with exacerbation of polymyositis with a second challenge9,19). But Halla et al (1984)20) suggested to readminister D-penicillamine after polymyositis, especially if an adequate interval of time has elapsed before administration of the drug is resumed.
Although polymyositis can develop as a complication of D-penicillamine therapy, polymyositis rarely has been reported to be associated with primary biliary cirrhosis. Only 9 cases of polymyositis associated with primary biliary cirrhosis have been reported21ŌĆō28). In 4 patients primary biliary cirrhosis antedated polymyositis by 1ŌĆō6 years, in 4 patients the diseases were diagnosed simultaneously, and only in one patient polymyositis preceded primary biliary cirrhosis. In our case polymyositis developed 5 years after primary biliary cirrhosis was diagnosed. Whether D-penicillamine treatment was the sole cause of polymyositis or acted as a trigger for the development of a secondary or an overlap type of autoimmune disease in the patient who already had a primary autoimmune disorder like primary biliary cirrhosis remains to be determined.
To our knowledge, this is the first case of polymyositis developed in a patient treated with D-penicillamine in Korea. Patients receiving D-penicillamine therapy should be followed carefully for the developmenht of autoimmune complications like polymyositis/dermatomyositis.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print





