


INTRODUCTION
Hepatocellular carcinoma (HCC) is frequently associated with hepatitis B virus (HBV) infection1,2). Aspects of HBV infection that possibly may contribute to the malignant transformation of hepatocytes include integration of HBV in the host genome3,4) and the production by HBV of transactivating proteins, such as the protein products of the X gene5–7) and a part of the pre-S2/S gene8,9). However, no specific pattern of HBV-DNA integration has been detected so far10,11) and no transforming oncogene has been discovered within the HBV genome12). Thus, the molecular mechanism of HBV-associated hepatocarcinogenesis is still poorly understood.
HBV containing a stop mutation of precore codon 28 has been isolated frequently from the serum of some HBV chronic carriers13–16). This mutant cannot produce HBeAg but retains its replicative capacity17). It has been reported often in association with the development of fulminant hepatitis14,18) and it has been found in some HCC patients19,20). It is possible that this mutant virus could contribute to the development of HCC. In the present study, HBV from the serum of HCC patients was analyzed to detect a stop mutation at precore codon 28 in an attempt to define further the possible role of such a mutation in hepatocarcinogenesis.
MATERIALS AND METHODS
1. Patients
Seventy-three HCC patients with hepatitis B surface antigen (HBsAg) and 19 controls with HBsAg (without HCC) were seen at the Department of Internal Medicine, Kangnam St. Mary’s Hospital, Catholic University Medical College, Seoul, Korea, between 1990 and 1993 (Table 1). All were negative for antibody to the hepatitis C virus (anti-HCV). Of the 73 HCC patients, the diagnosis of HCC was made histologically in 61 and was based on elevated levels of alpha-fetoprotein (AFP) (>400ng/ml) and CT scan in 12. Cirrhosis was visualized at laparoscopy in 46/61 (75%) HCC patients. Serum samples were collected at the time of the initial hospital admission and were stored at −20°C until tested in the present study. The 19 controls had histologic evidence of chronic active hepatitis and included 9 with HBeAg and 10 with antibody to HBeAg (anti-HBe) (HBeAg-negative).
2. Serological Tests
For patients and controls, sera were tested for HBsAg, antibody to HBsAg, HBeAg, anti-HBe and antibody to the hepatitis B core antigen using commercially available radioimmunoassays (Abbott Laboratories, North Chicago, IL, USA), and antibody to the hepatitis C virus (anti-HCV) using commercially available enzyme immunoassays (Abbott Laboratories).
3. DNA Extraction from Serum
One hundred microliters of each serum sample were mixed with 200ng of proteinase K solution (20mg/ml; Boehringer Mannheim, Mannheim, Germany) in 100 μl of 100mM NaCl, 10 mM Tris-HCl (pH 8.0), 25mM ethylenediaminetetraacetic acid (pH 8.0), and 0.5% sodium dodecyl sulfate, and incubated at 56°C for 2 hours. This was sequentially extracted with an equal volume of phenol, with an equal volume of a solution of phenol: chloroform: isoamyl alcohol (25:24:1), and with an equal volume of chloroform. The extracted sample was mixed with 3 μl glycogen and one-tenth volume of 3M sodium acetate (pH 5.2) and precipitated overnight with three volumes of cold 100% ethanol. The precipitated DNA was washed with 70% ethanol, air dried, and dissolved in 100 μl TE buffer (10mmol/L Tris-HCl and 0.1mmol/L EDTA, pH 8.0).
4. Polymerase Chain Reaction
The reaction mixture (100 μl) contained 50 μl of prepared DNA template, 10mmol/L Tris (pH 8.3), 50mmol/L KCl, 1.5mmol/L MgCl2, 1% (w/v) gelatin, 0.5 units of Taq polymerase (Perkin-Elmer; Norwalk, CT, USA), 200mmol/L each of the four deoxynucleoside triphosphates and 1 mmol/L each of sense primer (nucleotides 1689–1708; 5′-ACCTTGAGGCATACTTCAAA) and antisense primer (nucleotides 2058–2078; 5′-CAGAATAGCTTGCCTGAGTGC) for a DNA fragment of 390 bp, which includes the precore and proximal core regions of the HBV genome. The reaction mixture was overlaid with a drop of mineral oil, heated at 95°C for 5 minutes, and amplified with a programmable thermal cycler (Perkin-Elmer). Thirty-five amplification cycles were performed, each consisting of denaturation of template DNA at 95°C for 1 min, primer annealing at 55°C for 1 minute, and polymerization at 72°C for 2 minutes. After the last cycle, the temperature was maintained at 72°C for 10 minutes for complete polymerization. Ten microliters of each polymerase chain reaction (PCR) product were separated by electrophoresis in a 2% agarose gel and stained with ethidium bromide to visualize the amplified DNA fragment.
For sera with no detectable HBV DNA by the PCR process described above, DNA was re-extracted from 250 μl of serum and two PCR amplifications were performed. In the first step, amplification conditions were the same as above; in the second amplification step, 5 μl of the PCR product were amplified for 30 cycles using the same primers with primer annealing conditions at 60°C for 1 minute.
Care was taken to avoid cross-contamination between different steps in the procedure; preparation of the DNA template and preparation of the PCR mixture were conducted in a separate room from the amplification, gel electrophoresis, and purification of PCR products. All samples and solutions were handled with aerosol-resistant pipette tips, and fresh aliquots of reagent solutions were used for each experiment. Positive and negative controls were run with each amplification.
5. Sequencing
Sequencing of DNA amplified by PCR was performed according to the Sanger dideoxy chain termination method using a commercially available sequencing kit (“fmol”; Promega, Madison, WI, USA). The PCR product was purified using a microconcentrator (microcon-100; Amicon, Danvers, MA, USA). Ten pmol of a sequencing primer (nucleotide 1776–1797; 5′-GGCTGTAGGCATAAATTGGTC) for the sense strand of the precore and proximal core regions were end-labeled with 10 pmol of [γ-32P]-adenosine triphosphate (Amersham, Arlington Heights, IL, USA). The sequencing product was electrophoresed with 6% polyacrylamide/7 M urea gel, dried at 80°C for 1 hour with a gel drier, and autoradiographed at −70°C for 2–24 hours.
RESULTS
2. Stop Mutation of Precore Codon 28
Among those cases whose sequencing results were sufficient for analysis, 50/58 (86%) of the HCC patients and 12/16 (69%) of the controls with chronic hepatitis B had an HBV with a mutation at the 83rd nucleotide of the precore region (TGG →TAG) that changes codon 28 from tryptophan to a stop codon (Table 3).
3. Infection Patterns of Mutant- and Wild-type HBV and HBeAg
Among HCC patients, the precore codon 28 stop mutant was detected in 23/28 (82%) HBeAg-positive patients and 27/30 (90%) HBeAg-negative patients. Among controls with chronic hepatitis B, this mutation was detected in 5/9 (56%) HBeAg-positive controls and 7/7 (100%) HBeAg-negative controls (Table 4).
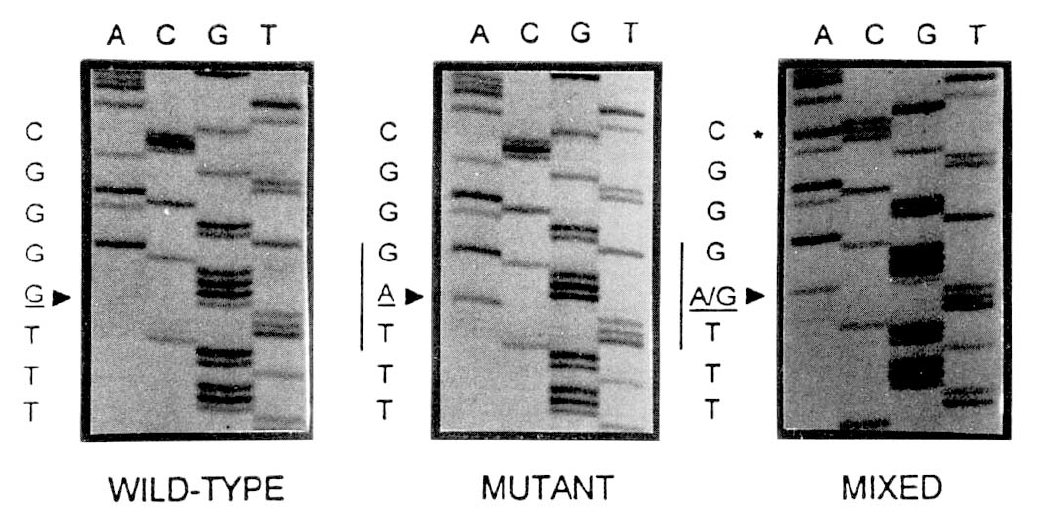
Of the 28 HBeAg-positive HCC patients, 5 (18%) had wild-type HBV alone, 13 (46%) had a mixed infection with both the wild-type and mutant HBV in their serum, since two bands could be detected at the second nucleotide of the precore codon 28 (Fig. 1), and 10 (36%) were infected with the mutant-type alone. Of the 9 HBeAg-positive controls, 4 (44%) had the wild-type alone and 5 (56%) had a mixture of both strains (Table 4).
Of the 30 HBeAg-negative HCC patients, 3 (10%) had wild-type HBV alone, 8 (27%) had a mixed infection with both strains and 19 (63%) were infected with the mutant-type alone. Of the 7 HBeAg-negative controls, 4 (57%) had a mixed infection of both strains and 3 (43%) were infected with the mutant-type alone (Table 4).
4. Precore Codon 28 Stop Mutation and Cirrhosis
Sequencing results from HCC patients were available for 50 of the 61 patients on whom laparoscopic examination was performed. Among those, cirrhosis was observed in 34/43 (79%) patients with the precore codon 28 stop mutant, including 22/24 (92%) with the mutant-type alone, compared to 12/19 (63%) HCC patients with a mixture of wild- and mutant-type HBV (p<0.03) (Table 5).
5. Precore Codon 28 Stop Mutation and Clinical Parameters
The 19 anti-HBe-positive HCC patients with mutant HBV alone were older (median age 53 years; range 37–64 years), compared to the 17 HBeAg-positive HCC patients with co-infection of both wild-type and mutant HBV (median age 43 years; range 23–66 years) (p<0.05). They also had lower serum aspartate aminotransferase levels (82±3IU/L vs. 192±75IU/L) (p<0.03) and lower serum gamma-glutamyl transpeptidase levels (107±2IU/L vs. 214±35IU/L) (p<0.03).
DISCUSSION
Among HCC patients with HBV-DNA, HBV with the precore codon 28 stop mutation was detected in 82% of HBeAg-positive and 90% of HBeAg-negative patients. Among chronic hepatitis B controls, 56% of the HBeAg-positive cases and 100% of the HBeAg-negative cases had the mutant virus. These results indicate that this mutant virus infection is frequently found both in patients with HBeAg and in patients who are HBeAg-negative.
A mixed infection with both the wild-type and the mutant HBV has been reported in 30–79% of HBeAg-positive patients with chronic hepatitis B21,22). Among HBeAg-positive patients in the present study, 46% of the HCC patients and 56% of the chronic hepatitis B controls had such a pattern, but the total prevalence of this mutant virus in the HBeAg-positive HCC patients (alone or with wild-type HBV, 82%) was much greater than that of the controls (56%). These observations suggest that HBV with the precore codon 28 stop mutation may appear in the early stages of HBV infection and may coexist with the wild-type virus prior to the development of HCC, although the actual order of events cannot be determined from the data reported here.
The data in this study suggest the presence of HBV with a stop mutation at precore codon 28 in HCC patients may not be closely correlated with the absence of HBeAg. Among HBeAg-positive HCC patients in the present study, 36% had only the precore codon 28 stop mutant without wild-type HBV; among HBeAg-negative HCC patients, 63% had mutant HBV alone. In contrast, 0/9 HBeAg-positive controls with chronic hepatitis B and 3/7 (43%) HBeAg-negative controls had the mutant type HBV alone.
In the present study, HCC patients with the mutant virus alone (frequently HBeAg-negative) were older in age and had lower serum levels of AST and GGT, compared to patients with a mixed infection of both the wild-type and the mutant HBV (frequently HBeAg-positive). Furthermore, patients with the mutant alone more often had cirrhosis than did patients with mixed infections. These clinical features support the suggestion that the HBV infection evolves from infection with the wild-type virus, to a mixed viral population of wild-type and mutant-type, and eventually to a population consisting of the mutant HBV alone in some patients22). This progression may possibly occur in association with worsening liver damage22), as in the association with cirrhosis in the present study. It is possible that the prolonged presence of this mutant replicative virus could play an active role in carcinogenesis.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print





