


INTRODUCTION
One of the major paradoxes in understanding the clinical effects of IL-2 remains the dichotomy between almost universal susceptibility of tumor cells to IL-2 activated lymphocytes (termed lymphokine activated killer or LAK cells) in virto and the low respnse rates observed in clinical trials1ŌĆō9). In the two most susceptible cancers, renal cell carcinomal and malignant melanoma, only 10ŌĆō25% response rates and 5ŌĆō10% complete response rates have been reported7ŌĆō9). These findings have raised the possibility that there may be inhibitory factors against LAK cell activation in vivo, which therefore diminish anti-tumor effects of IL-2 therapy.
NO is a well known cytotoxic effector molecule which may contribute to the development of cell mediated immune responses in a number of ways including tumor cell killing10,11). On the other hand, NO is potentially immunosuppressive, resulting in decreased lymphocyte proliferation and cytotoxic activity12ŌĆō15). Thus, both anti-tumor and tumor promoting activities of NO appear possible in vivo.
IL-2 therapy is known to induce synthesis of proinflammatory cytokines such as IFN╬│, TNF and IL-1 by LAK cells16,17). The same mediators are known to induce NO synthase expression in macrophages18). Hibbs and co-workers demonstrated that IL-2 treated patients developed marked (6ŌĆō10 fold) increases in NO synthesis, peaking on days 5ŌĆō7 following a 5 day course of high-dose intravenous bolus IL-2 treatment19). These observations have raised a question whether high output NO produced during IL-2 therapy acts as an anti-tumor effector mechanism or an inhibitory mechanism against the anti-tumor effect of IL-2 therapy by suppressing LAK cell activities.
We, therefore, developed a murine ascites tumor model to evaluate the role of NO synthesis during IL-2 treatment, and demonstrated that L-arginine: NO synthesis pathway is activated in the local tumor tissue itself as well as systemically, and that NO synthesized during IL-2 treatment may be an inhibitory factor against anti-tumor effects of IL-2 therapy.
MATERIALS AND METHODS
1. Animals
Specific pathogen free BALB/c mice (ages 6ŌĆō8weeks) were obtained from Harlan-Sprague Dawley (Indianapolis, IN) and housed at the Chonbuk National University Hospital Animal Care Facility. Mice were maintained under guidelines established by the Chonbuk National University Hospital Animal Care Committee, which also approved experimental protocols. Mice were age and sex matched at the onset of each experiment. All experiments were performed at least twice with highly concordant results.
2. Tumor Cell Lines
Meth A tumor (a gift from Dr. Lloyd Old, Memorial Sloan Kettering Cancer Center, NY) is a methylcholanthrene-induced spindle cell skin cancer arising in a BALB/c mouse20). Tumor cells were maintained by serial intraperitoneal passage in syngeneic BALB/c mice or by culture RPMI 1640 supplemented with 5% fetal calf serum (Hyclone Laboratories, Inc., Logan, UT), 100 units/ml penicillin G (Sigma Chemical Co., St. Louis, MO), 50 ╬╝g/ml streptomycin (Sigma) and 2 mM glutamine (Sigma) (working medium). The ascites variant of this tumor is extremely aggressive and is rapidly lethal to mice in 12ŌĆō18 days following injection of 2ŌĆō3├Ś106 cells. Less virulent strains of this tumor exist, that have been attenuated by long-term passage in cell cultures.
Mycoplasmal infection was excluded by periodic surveillance cultrues of supernatants derived from the tumor cell line grown in antibiotic free media on pleuropneumonia-like organism agar (PPLO agar, Baxter Health Care Corporation, McGaw Park, IL) supplemented with 10% horse serum (Hyclone), 25% yeast extract (Gibed BRL, Gaithersburg, MD) and 10 units penicillin G/ml.
3. Treatment of Meth A Ascites-Bearing Mice with Subcutaneous IL-2
At the beginning (day 0) of each experiment, normal BALB/c mice were injected intraperitoneally with 2├Ś106 Meth A tumor cells. Beginning on day 3, groups of 5 mice were treated with a 5 day course of IL-2 (180,000 IU subcutaneously every 12h for 5 days). The recombinant human IL-2 was a generous gift from Chiron Corporation, Emeryville, CA. Some mice were implanted with osmotic minipumps (Model 2001, Alza Corporation, Palo Alto, CA) containing 0.2 ml of 3.38 M NG-monomethyl-L-arginine (MLA; a NOS inhibitor)21). In previous studies, we have demonstrated that these infusions block both baseline and cytokine-induced NO synthesis for 8ŌĆō9 days21).
4. Analysis of NO Synthesis by Ascites Cells
Ascites cells were cultured in working medium (7.5├Ś105 cells/ml) in 96 well flat bottom plates(at a final volume of 0.2 ml/well). After a 48h incubation at 37┬░C in humidified 95% air/5% CO2 atmosphere, nitrite, a stable metabolite of NO, was quantifed in culture supernatants using a previously described colorimetric microtiter assay21). Briefly, 50 ╬╝l samples of cell culture supernatant from each well were dispensed into flat bottomed microtiter plates, and incubated with 100 ╬╝l of a 1:1 mixture of 1% sulfanilamide in 30% acetic acid with 0.1% N-(1-naphthyl) ethylenediamine dihydrochloride in 60% acetic acid. Once mixed, this reagent was used within 5 min. Following a 5 min incubation with experimental samples or standards, absorbance at 570 nm was evaluated in a microtiter plate reader. Concentrations were determined from a linear standard curve generated from 6.25ŌĆō100 ╬╝M sodium nitrite in working medium. Results are presented as mean┬▒SD. Working medium contained < 0.5 ╬╝M nitrite.
6. Effect of Nitric Oxide Synthase Inhibition on IL-2 Induced Anti-Tumor Responses
Four groups (untreated control, MLA therapy only, IL-2 therapy only, IL-2/MLA therapy groups) of 5 normal BALB/c mice were acclimated to a nitrate/nitrite free diet (Ziegler Brothers, Gardeners, PA) in metabolic cages (Nalgene, Rochester, NY). Following a 7 day acclimation (on day 0), mice were injected intraperitoneally with 2├Ś106 Meth A tumor cells. On day 2, two groups (MLA therapy only and IL-2/MLA therapy groups) of mice were implanted with subcutaneous Alzet continuous infusion pumps (Model 2001) containing 0.2 ml of 3.38 M MLA. The other groups of mice underwent subcutaneous tunnel formation but no pump implantation. On day 3, IL-2 therapy (180,000 IU s.c. every 12h for 5 days) was begun in IL-2 therapy only and IL-2/MLA therapy groups. Daily body weight measurement and survival of the mice were evaluated to assess therapeutic responses. The concentration of nitrate in sequential 24 hour urine samples was measured using a previously described method21). Briefly, 50 ╬╝l of urine samples were dispensed into triplicate wells of round bottom microtiter plates and incubated for 90 min with 50 ╬╝l of a frozen suspension of Escherichia coli, induced for the enzyme nitrate reductase22). Following this incubation, plates were centrifuged at 1,000 xg for 10 min and 50 ╬╝l of supernatant were transferred to clean flat bottom microtiter plates and analyzed for nitrite as described previously.
RESULTS
1. Subcutaneous IL-2 Administration Induces NO Synthesis by Cells in Meth A Ascites
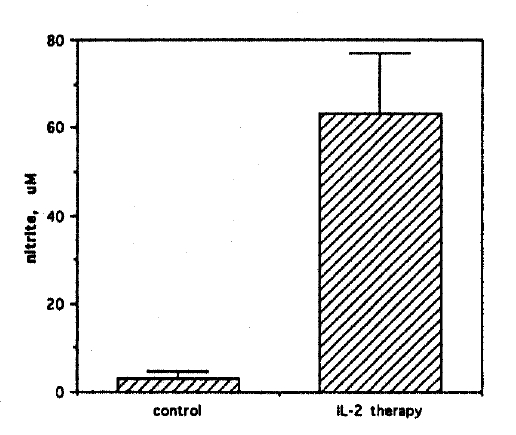
Cellular ascites from Meth A bearing mice were harvested under sterile conditions on day 8 following the last dose of IL-2. Following erythrocyte lysis, cells were cultured at 1.5├Ś105 cells/well in 0.2 ml working medium for 48h and analyzed for nitrite synthesis. Ascites cells dervied from untreated mice served as controls. Significant nitrite accumulation (63┬▒14 ╬╝M) was observed in supernatants of Meth A ascites cells from IL-2 treated mice (Fig. 1). In contrast, ascites cells from control mice synthesized little nitrite (3.2┬▒1.5 ╬╝M). This result demonstrated that nitrite synthesis was strongly induced in malignant ascites following subcutaneous IL-2 administration in mice. Further experiments were performed to confirm the origin of the nitrite from nitric oxide synthase.
2. Effect of MLA on NO Synthesis of Meth A Ascites Cells from IL-2 Treated Mice
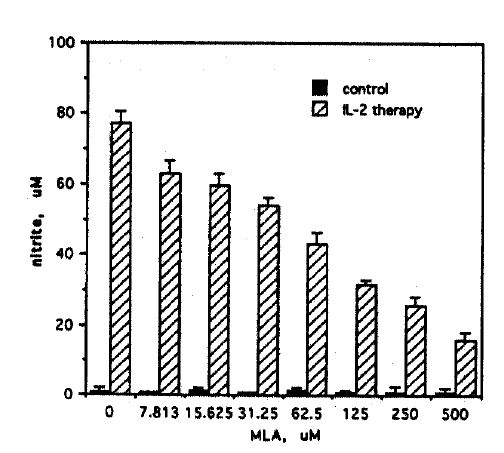
The nitric oxide synthase inhibitor MLA was used to establish that the source of nitrite in cell cultures was L-arginine: NO pathway. Ascites cells from IL-2 treated mice were cultured with increasing concentrations (0ŌĆō500 ╬╝M) of MLA. Nitrite production in these cultures was found to be inhibited in a dose dependent manner (Fig. 2). In contrast, MLA had no effect on the trace amounts of nitrite produced by ascites cells from non-IL-2 treated control mice (< 0.5 ╬╝M). Control experiments performed in the presence of 10 ╬╝g/ml polymyxin B gave similar results, excluding inadvertent endotoxin contamination.
Furthermore, bacterial lipopolysaccharide is undetectable in our working medium by Limulus lysate assay (sensitivity, 100 pg/ml). This result demonstrated that the nitrite in ascites cell cultures from IL-2 treated mice was a product of the cytokine induced L-arginine: NO pathway.
3. Effect of NO Synthesis Inhibition on IL-2 Induced Anti-Tumor Responses in vivo
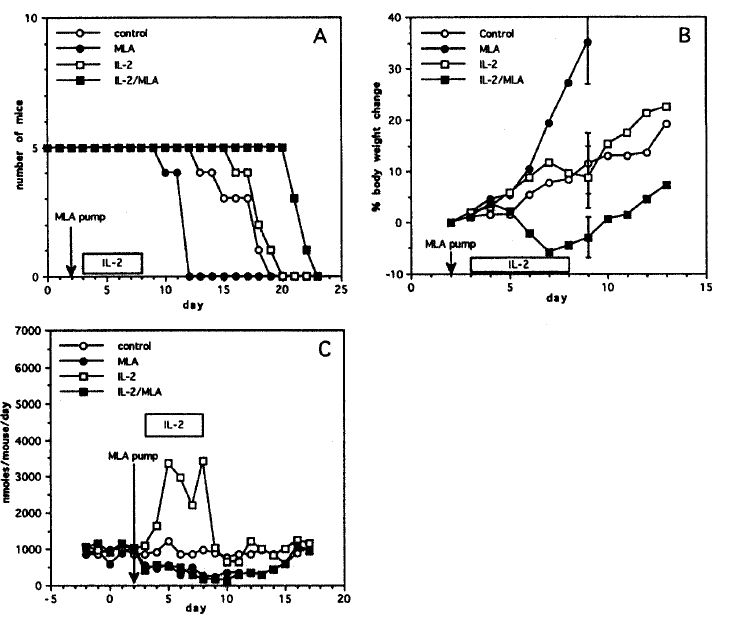
To evaluate whether IL-2 induced NO synthesis represents an anti-tumor effector mechanism or an inhibitory factor against therapeutic effects of IL-2, mice were acclimated to metabolic cages on a nitrate/nitrite-free diet. On day 0, four groups of mice (5 mice in each group) were injected intraperitoneally with 2├Ś106 Meth A tumor cells. On day 2, mice receiving MLA treatment were implanted subcutaneoulsy with Alzet continuous infusion pumps (Model 2001) containing 0.2 ml of 3.38 M MLA. The remaining groups of mice underwent anesthesia and subcutaneous tunnel formation but no pump implantation. On day 3, IL-2 therapy was begun (180,000 IU s.c. every 12h for 5 days). This experiment demonstrated that IL-2 therapy of Meth A ascites-bearing mice induced no significant changes in the rate of body weight increment (ascites growing) (on day 9, 8.8┬▒6.1% in IL-2 therapy group versus 11.5┬▒6.2% in control mice, p=0.365) and in mean survival (18.2 days in IL-2 therapy group versus 16.6 days in control mice, p=0.255) (Fig. 3A, 3B). In contrast, chronic MLA administration significantly decreased the rate of body weight increment (on day 9, ŌłÆ2.9┬▒5.0% in IL-2/MLA therapy group versus 8.8┬▒6.1% in IL-2 therapy group, p < 0.001) and prolonged mean survival of IL-2 treated mice (21.8 days in IL-2/MLA therapy group versus 18.2 days in IL-2 therapy groups, p=0.001).
Interestingly, the MLA treatment increased the rate of body weight increment (on day 9, 11.5┬▒6.2% in control group versus 35.1┬▒7.2% in MLA therapy group, p < 0.001) and diminished the survival of untreated control mice (16.6 days in control group versus 11.6 days in MLA therapy group, p=0.003). In order to confirm the effectiveness of NOS inhibition, pooled daily urines from experimental mice were analyzed for urinary nitrate excretion (Fig. 3C). MLA administration via Alzet continuous infusion pumps achieved approximately 60% suppression of urinary nitrate excretion by control mice. Subcutaneous IL-2 treatment strongly induced nitric oxide synthesis (up to 3.5 ╬╝moles/mouse/day), a 4-fold increase. MLA effectively suppressed NO production due to cytokine stimulation when given as subcutaneous continuous infusion.
DISCUSSION
NO is a well known cytotoxic effector molecule which may contribute to the development of cell mediated immune responses in a number of ways including the primary defense against facultative and obligate intracellular pathogens23) and tumor cell killing10,11). On the other hand, NO has potentially immunosuppressive effects on anticancer immune responses. A large number of studies have demonstrated that macrophage NO synthesis can decrease lectin and antigen stimulated lymphocyte proliferation12ŌĆō15). In high concentration, NO appears to inhibit antigen-specific cytotoxic T cell activation during infection14,15) and during the course of graft-versus host disease24,25). NO may also decrease macrophage Class II MHC antigen expression, which may impede the process by which antigen is presented to T cells26). Our own studies have established that direct exposure of splenocytes to high concentrations of NO strongly inhibits IL-2 induced LAK cell activation and expansion (manuscript in preparation). Thus, both anti-tumor and tumor promoting activites of NO appear possible in vivo.
The observation that MLA is a potent competitive inhibitor of the NOS enzyme provided an additional experimental tool to differentiate effector functions mediated by the L-arginine: NO pathway27,28).
LAK cell induction is observed in most IL-2 treated patients29,30) and has been thought to be important in mediating anti-tumor responses of IL-2 therapy. Hibbs and co-workers recently demonstrated that IL-2 treatment of patients with malignant melanoma and renal cell carcinoma strongly induces NO synthesis19). We hypothesized that cytokine induced NO synthesis represented a novel inhibitory factor against LAK cell activation and therefore diminished the anti-tumor effect of IL-2 therapy. In the present study, we employed Math A ascites tumor, which is relatively resistant to IL-2 therapy, as a model. Subcutaneous injections of IL-2 into a remote site were used to produce a systemic immune response. This mode of treatment avoided recurrent intraperitoneal injections, that could have disseminated ascites tumor.
Experiments reported herein established that significant nitrite synthesis was induced in malignant ascites during IL-2 treatment. Addition of the NO synthase inhibitor MLA to cell cultures demonstrated a dose dependent inhibition of the nitrite synthesis, confirming the origin of the nitrite to be from the L-arginine: NO pathway. These results strongly indicate that NO synthesis can be induced in ascites tumor cells by IL-2: This experimental result is consistent with earlier observation by Cox et al. who demonstrated that IL-2 could activate murine macrophage NO synthesis, particularly in the presence of IFNg31). Further studies will be necessary to establish whether IL-2 is acting directly on macrophages, via IL-2 receptors32). Alternatively, secondarily released inflammatory mediators, such as TNF, IL-1 and IFNg, poroduced by lymphocytes within the ascites, may play an obligate role in IL-2 induced NO synthesis by macrophages31,33). Cytokine-activated macrophages are known to increase secretion of IL-1, TNF and IL-6, which may also enhance induction fo iNOS activity within macrophages themselves and tumor cells34,35). The cellular source of NO produced during IL-2 therapy remains to be studied. Tumor infiltrating macrophages and tumor cells may be the first candidates for the NO synthesis because there are precedents for macrophages and tumor cells to synthesize NO following cytoking induction10,11,34,35). It seems unlikely that T or NK lymphocytes contribute significantly to intraperitoneal NO synthesis. Our unpublished data represented that the cytokine exposure, which are well known stimulus to iNOS expression in macrophages10,11,19), did not induce NO synthesis in lymphocyte cultures.
This model demonstrated that IL-2 treatment marginally increased mean survival of tumor bearing mice from 16.6 to 18.2 days (p=0.225). The subcutaneous regiment of IL-2 administration induced marked (up to 4 fold) increases in urinary nitrate excretion, demonstrating potent induction of NO synthesis in vivo. This increase was markedly inhibited by concomitant MLA administration. At the same time, the marginally beneficial effects of IL-2 treatment on survival was significantly increased by MLA administration, prolonging mean survival from 18.2 to 21.8 days (p=0.001). Likewise, the rate of body weight increment of IL-2 treatment group was significantly delayed by the treatment with MLA. These results indicated that high output NO synthesized during IL-2 therapy is an inhibitory factor against the therapeutic anti-tumor responses. In contrast, survival and the rate of body weight increment of tumor-bearing control mice were diminished and increased respectively by MLA administration, suggesting the possiblity of small but definite anti-tumor effect of baseline NO synthesis in these animals. The latter finding is consistent with earlier observations by Yim et al. who demonstrated that endogenous baseline NO production in mice bearing antigenic subcutaneous tumor decreases the rate of tumor progression21,37).
Mechanisms which can explain the contrasting difference of the role of NO in tumor immunology between the absence and presence of IL-2 therapy remain to be clarified. To unify these conflicting series of observations, we have proposed that, in the absence of IL-2 therapy, there may be little or no activated cytotoxic lymphocytes in tumor tissues and lymphocytes may, therefore, not be a major anti-tumor effector mechanism. Baseline endogenous NO synthesis may then be an important mechanism which delays tumor growth in this setting. In contrast, in the presence of IL-2 thereapy, both LAK cell activation, as well as high output NO synthesis, are induced in the tumor microenvironment. This high output NO can be cytotoxic not only to tumor cells but also prominently inhibit induction of LAK cell activation and proliferation. Based on the previous reports indicating that the anti-tumor effects of NO are mainly cytostatic rather than cytolytic11), but those of LAK cells are cytolytic rather than cytostatic1ŌĆō6), it may be possible that LAK cells are more effective than high output NO in inhibiting tumor growth. Inhibition of NO synthesis during IL-2 therapy, may therefore, result in increases in LAK cell activation and be beneficial to the therapeutic anti-tumor responses. The interrelationship of the anti-tumor and immunosuppressive roles of NO is likely to be complex and will require further evaluation.
Systemic high output NO synthesis is very likely to contribute to dose limiting toxicities of IL-2 therapy, such as hypotension, vascular leak and decreased myocardial performance (Yim, et al., manuscript submitted). Thus, the possibility exists that MLA administration during IL-2 therapy is beneficial in preventing the toxicities, as well as in promoting therapeutic anti-tumor responses.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print





