


INTRODUCTION
Primary pigmented nodular adrenocortical disease (PPNAD) is a rare cause of Cushing’s syndrome and occurs in younger age group (infants, adolescent, and young adults) than classic pituitary dependent Cushing’s disease. It’s characteristic symptoms result from hypersecretion of adrenal corticosteroid independent of pituitary adrenocorticotropic hormone. The symptoms of hypercortisolism are frequently mild, and a long interval between the onset of symptoms and the diagnosis is common. PPNAD occurs sporadically or as part of a familial syndrome called Carney complex1). Biochemically, it is characterized by hypercortisolism resistant to dexamethasone suppression, and pathologically characterized by small to normal sized adrenal gland, multiple small black pigmented nodules and internodular cortical atrophy.
Recently we experienced a case of Cushing’s syndrome due to primary pigmented nodular adrenocortical disease in a 44-year-old Korean woman and report this case with reviews of the literaiture.
CASE REPORT
A 44-year-old woman was admitted to our hospital for the evaluation of Cushing’s syndrome. She was diagnosed as hypertension at a local clinic in 1986 but recieved no antihypertensive medication. She developed headache, nausea and vomiting in Feb, 1990 and was admitted to the department of neurosurgery with diagnosis of Rt. cerebellar hemorrhage. After discharge, she has been followed for treatment of hypertension. During the follow-up period, she was found to have the Cushingoid features and transferred to the Department of Internal Medicine for the evaluation of Cushing’s syndrome.
The family and other past medical history were not contributory.
On admission, blood pressure was 150/110 mmHg, pulse rate 78/min, body temperature 36.5°C, respiration rate 18/min and height was 164cm and body weight was 77kg.
On physical examination, she was shown to have central obesity. Her face was round and plethoric. She had pink conjunctivae and white sclerae.
On auscultation of the chest, breathing sounds were normal and the heart sound was regular without murmur. On examination of the abdomen, she was found to have striae. Further examination revealed no abnormal finding.
Laboratory studies included hemoglobin 12.7g/dl, WBC 6700/mm3 with 75% neutrophils and 23% lymphocytes, platelet 261,000/mm3, total protein 7.2g/dl, albumin 4.2g/dl, total bilirubin 0.7mg/dl, direct bilirubin 0.3mg/dl, ALT 17unit, AST 19 unit, alkaline phosphatase 65U/L, BUN 20mg/dl and creatinine 1.1mg/dl. The concentrations of Na, K, CI and Ca were normal. FBS was 116mg/dl and HbA1c was 3.1%. Urinalysis was normal.
The results of low and high dose dexamethasone suppression test were shown in Table 1.
Chest PA and skull series revealed no abnormal findings.
By her history and physical examination, we suspect she had Cushing’s syndrome and then measured 24hr urine free cortisol and performed low dose dexamethasone suppression test. After this study, high dose dexamethasone suppression test was done to differentiate the etiology of Cushing’s syndrome. The levels of plasma cortisol were not suppressed and plasma ACTH level was very low. Abdominal CT scanning was done under the impression of adrenocortical tumor but revealed no abnormal mass in the adreanal gland first. Therefore we performed pituitary fossa MRI to rule out Cushing’s disease associated with micronodular adrenal hyperplasia. MRI reveals atrophic pituitary gland with empty sella. At that time, we didn’t decide the treatment modality for this patient because of poor general condition.
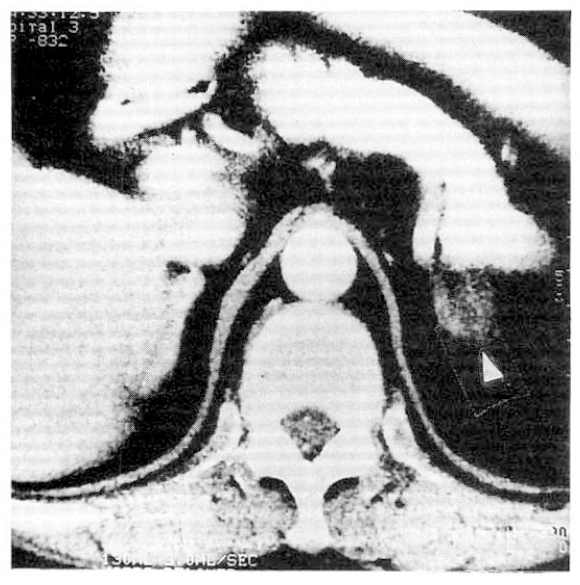
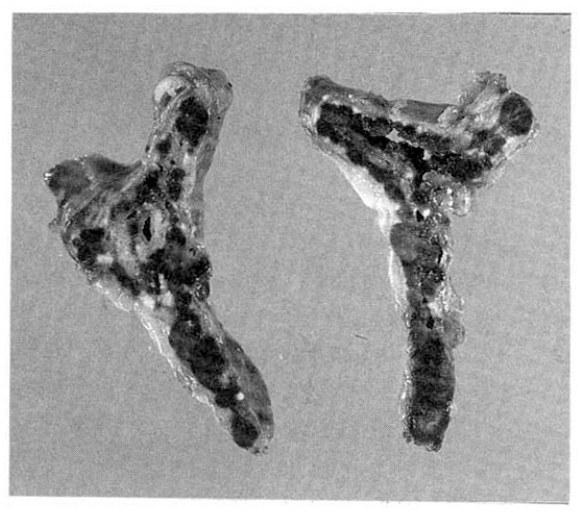
Instead, follow-up study was recommended. Follow-up abdominal CT scan, 3 years after the first study, revealed 2cm-sized hypodense mass in the upper pole of lt, adrenal gland (Fig. 1). So, explorative laparotomy was done under the impression of lt, adrenal adenoma. At operation field, no abnormal adrenal mass was found but the surgeon performed lt, adrenalectomy. The cut surface of surgical specimen showed multiple black pigmented nodules with pale atrophic cortex (Fig. 2).
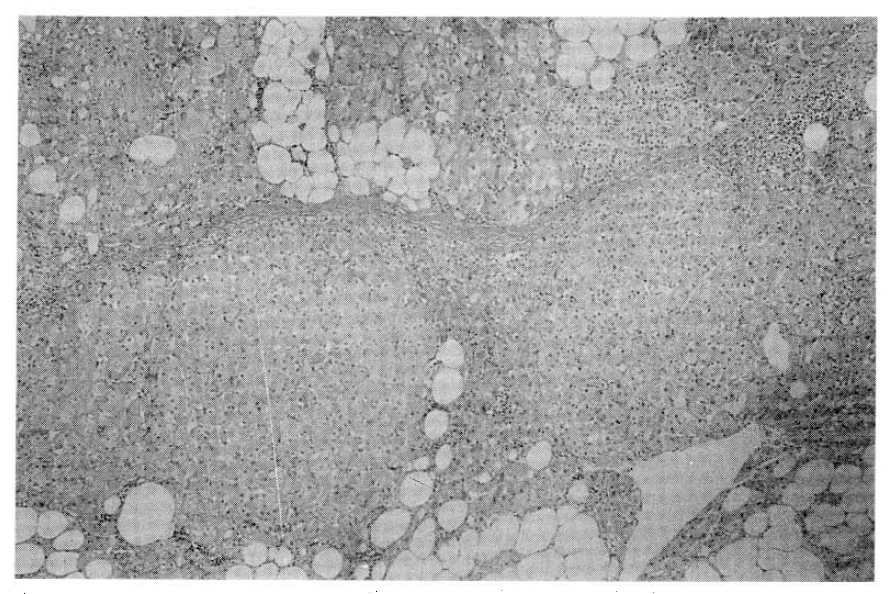
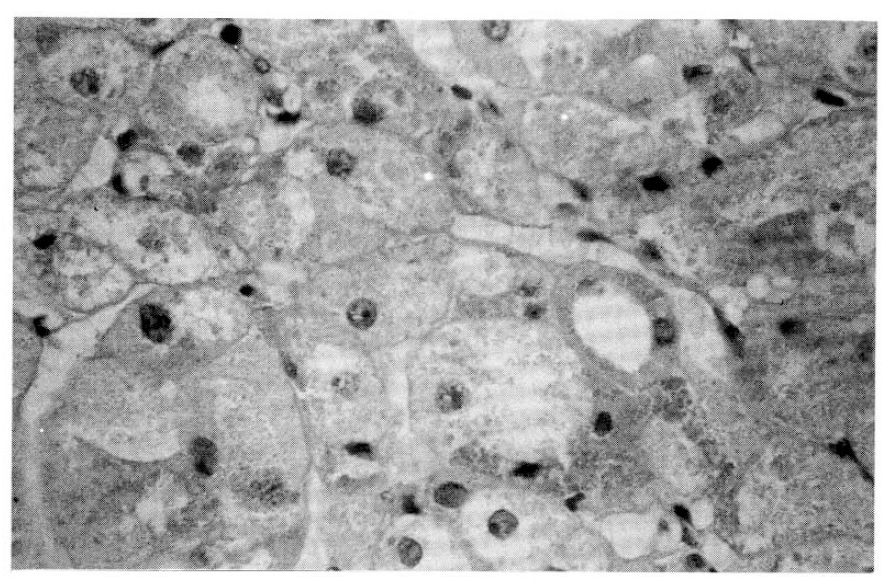
On the microscopic finding, pigmented micronodular dysplasia with cortical atrophy was found (Fig. 3, 4).
After operation, her blood pressure was normalized, and follow-up hormonal levels were also improved.
We concluded that her Cushing’s syndrome resulted from primary pigmented nodular adrenocortical disease and we plan the contralateral adrenalectomy.
DISCUSSION
The primary cause of the Cushing’s syndrome may be in the adrenal gland (hyperplasia or neoplasm or infrequently primary pigmented nodular adrenocortical disease like the above patient), outside of adrenal (pituitary adenoma or ectopic secretion of ACTH), iatrogenic (administration of adrenocorticosteroid) (Table 2).
PPNAD is a rare cause of Cushing’s syndrome in infants, children and young adults. It is characterizd by non-adrenocorticotropic hormone dependent hypersecretion of cortisol by multiple, pigmented nodules of hyperplastic adrenocortical cells.
The above patient presents a rare form of primary adrenal disease producing Cushing’s syndrome that is ACTH-independent. The low plasma ACTH level, lack of suppression with high-dose dexamethasone and the normal adrenal weight with atrophy between nodules all support this.
PPNAD may be familial and associated with cardiac myxomas, spotty skin pigmentation (lentigines), myxomatious tumors of the breast and soft tissues and large cell calcifying Sertoli cell tumors of the testes (Carney complex)2–5).
The pathogenesis of the disease is unkown, but some data suggest that it may be due to circulating adrenal-stimulating immunoglobulins6–8).
Munro et al proposed autoimmune stimulation of the adrenal cortex as a cause of adrenal hyperfunction. Such stimulation of the adrenal cortex would be analogous to the stimulation of thyroid tissue by thyroid – stimulating immunoglobulins in Graves’ disease9).
Biochemically, PPNAD is Characterized by elevated levels of plasma and urinary cortisol that are not suppressed by high dose dexamethasone.
Plasma adrenocorticotropic hormone (ACTH) levels are low or undetectable, both peripherally and in selectively sampled inferior petrosal sinses, and ACTH levels do not respond to an intravenous injection of corticotropin releasing hormone (CRH).
Pathologic findings are character. 1) decreased, normal, or slightly increased total gland weight and both involved; 2) studding of the external and cut surfaces by small (<4mm) black, brown nodules; (The pigment is lipofuschin) 3) The nodules are unencapsulated and composed or closely packed polyhedral cells; 4) The intervening cortical tissue is atrophic, thih distinguishes these glands from the nodular hyperplasia of pituitary-dependent Cushing disease, in which the cortical tissue between the nodules is hyperplastic10–12), invasion into pericapsular fat and vascular structures described13). However, no evidence of metastasis or associated adrenal carcinoma has been described.
Radiologically, osteoporosis is frequently seen on bone film in Cushing’s syndrome due to PPNAD. Computed tomographic scanning of pituitary shows no abnormalities and computed tomographic scanning of adrenal glands may show normal or slightly enlarged glands without evidence of mass. Doppman et al14), suggest that the detection of multiple, small adrenocortical nodules with CT or MRI supports the diagnosis of PPNAD.
The importance of recognizing this diagnosis, as a separate condition, is to identify the necessity for bilateral adrenalectomy to cure the cushingoid symptoms. This is demonstrated by the failure of pituitary irradiation or unilateral adrenalectomy to control the symptoms15,16) because of recurrence. The absence of any identified Nelson’s syndrome following adrenalectomy, and the lack of dexamethasone suppressibility, indicate adrenal autonomy.
After unilateral adrenalectomy, our patient’s blood pressure was normalized and biochemically cortisol level also was lowered. We think it may be due to the predominant hyperplasia of lt, adrenal gland compared to the right side.
But we are planning contralateral adrenalectomy because Cushing’s syndrome will be recurred during follow-up.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print





