


INTRODUCTION
Quinupristin/Dalfopristin (Synercid┬«), a combination of two different streptogramins antibiotics, has a bactericidal effect by inhibiting the bacterial protein synthesis of ribosome1ŌĆō4). It is currently used in infections caused by Vancomycin-resistant Enterococcus faecium (VREF), when no other active antibacterial agent is available1). The known adverse effects of Quinupristin/Dalfopristin are peripheral vasculitis, arthralgia, myalgia, nausea, diarrhea, vomiting and skin papules2, 3, 5).
SweetŌĆÖs syndrome, or acute febrile neutrophilic dermatosis, is characterized by painful skin papules, and plaques, fever and peripheral blood leukocytosis, with dermal neutrophilic infiltration on a skin biopsy6, 7).
Drug-induced SweetŌĆÖs syndrome has a temporal relationship between the drug ingestion and clinical presentation, or a temporally-related recurrence after oral rechallenge, with temporally-related resolution of the lesions following drug withdrawal or on treatment with systemic corticosteroids8ŌĆō13).
The authors, having encountered a patient, who was the first to our knowledge, developed SweetŌĆÖs syndrome after administration of Quinupristin/Dalfopristin, herein report on the case with a review of the English literature relating to the other adverse effects of the drug and the clinical features of the syndrome.
CASE REPORT
A 63-year-old woman, with end stage renal disease, receiving hemodialysis three times a week, was hospitalized for acute subdural hemorrhage. On the 30th day following her admission, she had a high fever of 39°C and complained of costovertebral angle (CVA) tenderness on physical examination. Chest radiographs showed no abnormalities. VREF was isolated in a urine culture, but with blood cultures yielding no growth of microorganism. Acute pyelonephritis, due to VREF, was diagnosed. The patient was treated with Quinupristin/Dalfopristin (Synercid®, 7.5 mg/kg, intravenously, through a central venous catheter, every 8 hours).
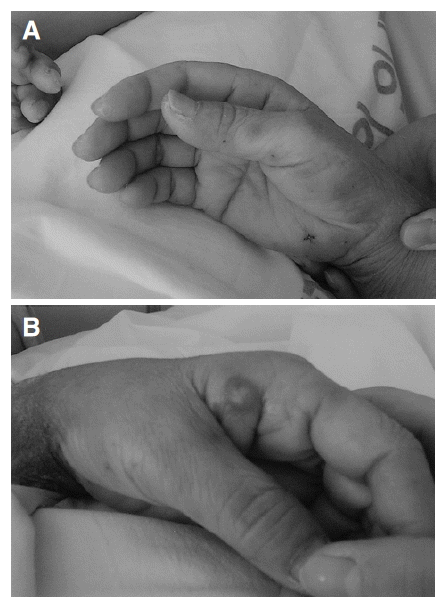
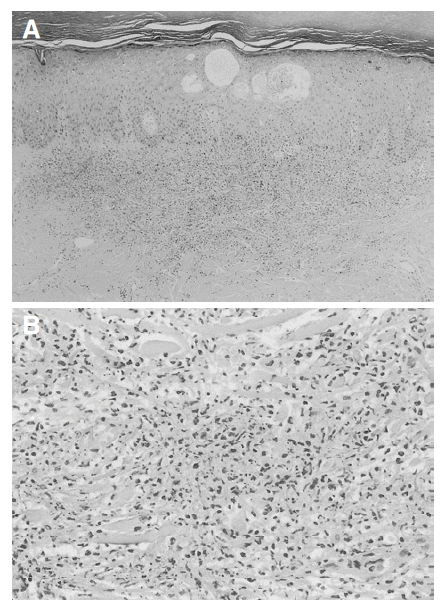
On the 1st day of the Quinupristin/Dalfopristin treatment, her body temperature was normalized and the CVA tenderness subsided, but she complained of nausea, vomiting, and myalgia. On the 2nd day of treatment with the drug, she complained of severe myalgia, arthralgia and general weakness, and presented tender erythematous 2 cm-sized papules on the palms of her hands (Figure 1A). On the 3rd day of treatment the skin papules, which started on the palm, spread to the face, chest and neck, and progressed to maculopapular plaques or pustules (Figure 1B). Her body temperature again rose to 38.7┬░C. The white blood cell count was 20,600 cells/mm3 (neutrophils, 84%), the hemoglobin was 7.0 g/dL, the platelet count was 209,000 cells/mm3, but her liver enzymes were within normal limits. There was no evidence of infections on physical examinations. Blood and urine cultures revealed no growth of microorganisms. A punch biopsy of a skin lesion on her palm was performed, which showed intra- and sub-corneal pustular formation, with epidermal leukocytosis (Figure 2). There was no evidence of vasculitis, necrosis or infections caused by bacteria, fungus or mycobacteria, in the biopsy specimen. These findings were compatible with SweetŌĆÖs syndrome. She had received no other drugs that could have caused the drug-induced SweetŌĆÖs syndrome.
The Quinupristin/Dalfopristin treatment was stopped, after which, the severity of these side effects were rapidly relieved, and the pain from the skin lesions subsided without analgesics. The maculopapular plaques and pustules, also, spontaneously disappeared. The 3-month follow-up revealed no recurrence of the disease.
DISCUSSION
In 1964, Sweet described a new disease, in eight female patients, which was characterized by the acute onset of fever, leukocytosis, and tender skin plaques infiltrated by neutrophils: he named the disorder acute febrile neutrophilic dermatosis6). In as many as 50 percent of patients, SweetŌĆÖs syndrome is associated with a variety of underlying diseases:7, 14, 15) malignancy, bacterial and viral infections, drugs (furosemide8), hydralazine9), oral contraceptives10), minocycline11), nitrofurantoin12), trimethoprim-sulfamethoxazole13) and Granulocyte-stimulating factor16), autoimmune and collagen vascular diseases (rheumatoid arthritis, systemic lupus erythematosus, mixed connective tissue disease, Hashimoto thyroiditis, SjogrenŌĆÖs syndrome), inflammatory bowel disease (ChronŌĆÖs disease, ulcerative colitis) and pregnancy.
The pathogenesis of SweetŌĆÖs syndrome is still unknown, but it is currently thought that cytokine dysregulation, a type 3-hypersensitivity reaction or a T cell-dependent cellular immune reaction is involved8ŌĆō13, 15).
Diagnostic criteria have been proposed for SweetŌĆÖs syndrome7), with a patient having to meet two major and two minor criteria for the diagnosis to be confirmed. One major criterion is the abrupt onset of typical cutaneous lesions, with the other being a histopathology compatible with SweetŌĆÖs syndrome. There are four minor criteria antecedent fever or infection; accompanying fever, arthralgia, conjunctivitis, or underlying malignancy; leukocytosis and good response to systemic corticosteriods, but no response to antibiotics15). The cutaneous lesions are characterized by tender or painful erythematous or violaceous plaques or nodules. They are most often found on the face, neck and upper extremities, especially on the dorsum of the hands, but they can occur anywhere7, 13, 15). The diagnostic histopathologic feature of SweetŌĆÖs syndrome is nodular and dense perivascular neutrophilic infiltration, with no evidence of vasculitis. The neutrophils can transepidermally migrate, creating subcorneal pustules6, 7, 14, 15). Drug-induced SweetŌĆÖs syndrome is found to be resolved promptly, without recurrences, on discontinuation of medication, or with systemic steroid therapy8ŌĆō13). In patients with drug-induced SweetŌĆÖs syndrome, neutrophilia is often absent13). It is difficult to definitively determine whether the cause of the dermatosis is drug-induced, or related to an underlying condition13). Two useful criteria for establishing a diagnosis of drug-induced SweetŌĆÖs syndrome are a temporal relationship between the drug administration and the clinical presentation, and between drug withdrawal and resolution of the disease.
VREF infections have recently become a particular public health concern. These organisms were first described, in Europe, in the mid-1980s1). Quinupristin/Dalfopristin is a novel parenteral antimicrobial agent, consisting of two different semisynthetic, water-soluble, streptogramins antibiotics, Quinupristin (30%) and Dalfopristin (70%). They are synergistic, and usually bactericidal against Gram-positive, selected Gram-negative and some anaerobic organisms, and is an effective therapy for serious infections due to VREF2). It has a unique mechanism of action that involves bacterial cell entry, by diffusion of the drug to the bacterial ribosome, followed by the formation of stable Dalfopristin-ribosome-Quinupristin complexes. This results in the irreversible inhibition of bacterial protein synthesis and bacterial cell death. Consequently, this combination of the two components may prevent the appearance of strains resistant to either of the components3, 4).
However, parenteral administration of Quinupristin/Dalfopristin can cause systemic adverse effects. Arthralgia, without objective signs of articulopathy, and myalgia are the most commonly reported adverse events, and the leading causes of treatment discontinuation. Gastrointestinal adverse events (nausea, vomiting, and diarrhea) and skin rash have also been documented2, 3, 5). All theses side-effects are reversible on discontinuing the drug, and its overall tolerability is good2, 5). Patients receiving at least one intravenous infusion of quinupristin/dalfopristin have been known to experience local venous intolerability (pain, burning, inflammation, and thrombophlebitis)2). Laboratory findings of the adverse events are the increases in alkaline phosphatase, aminotransferases and ╬│-glutamine transferase, as well as decreases in sodium, hemoglobin, haematocrit, platelet count and red blood cell count2, 3, 5)
The drug is primarily excreted by the liver (75%), with a small portion of the unchanged (15ŌĆō19%) form eliminated by the kidneys3, 17). A recent study indicates that no formal dosage reduction would appear necessary in patients with severe chronic renal impairment or hepatic dysfunction, although there is a tendency for slower clearance of unchanged dalfopristin and active quinupristin derivatives3, 17). However, the effect of a dose reduction or an increase in the dosing interval, on the pharmacokinetics of Quinupristin/ Dalfopristin in these patients requires further study, so no recommendations can be made at this time3).
Our patient, who received Quinupristin/Dalfopristin treatment, initially presented with arthralgia, myalgia, nausea, vomiting and fever, with maculopapular plaques that progressed to pustules. There was no evidence of infections or vasculitis. A skin biopsy revealed diffuse infiltration of neutrophils into the epidermis. After discontinuation of the drug, the systemic adverse events were quickly resolved, and the cutaneous lesions cleared within 7 days. The patient receive no systemic corticosteroids. Although unproved, it is not inconceivable that in this patient the Quinupristin/Dalfopristin was involved in the clinical and histological reactions. All theses findings were suggestive of Quinupristin/Dalfopristin-induced SweetŌĆÖs syndrome. The drug has never been implicated in the literature as a causative agent of SweetŌĆÖs syndrome before. Additional studies are warranted to determine the mechanism of SweetŌĆÖs syndrome associated with the drug.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print





