


INTRODUCTION
Therapeutic drug monitoring (TDM) is generally defined as the clinical laboratory measurement of a chemical parameter that, with appropriate medical interpretation, will directly influence drug prescribing procedures [1]. Otherwise, TDM refers to the individualization of drug dosage by maintaining plasma or blood drug concentrations within a targeted therapeutic range or window [2]. By combining knowledge of pharmaceutics, pharmacokinetics, and pharmacodynamics, TDM enables the assessment of the efficacy and safety of a particular medication in a variety of clinical settings [3-7]. The goal of this process is to individualize therapeutic regimens for optimal patient benefit. Traditionally, TDM involves measuring drug concentrations in various biological fluids and interpreting these concentrations in terms of relevant clinical parameters. Clinical pharmacists and pharmacologists use pharmacokinetic principles to assess these interpretations. The science of TDM introduced a new aspect of clinical practice in the 1960s with the publication of initial pharmacokinetic studies linking mathematical theories to patient outcomes [3]. From there, clinical pharmacokinetics emerged as a discipline in the late 1960s and early 1970s. Pioneers of drug monitoring in the 1970s focused on adverse drug reactions and demonstrated clearly that by constructing therapeutic ranges, the incidence of toxicity to drugs such as digoxin [8], phenytoin, lithium, and theophylline [9] could be reduced [10]. The emergence of clinical pharmacokinetic monitoring was encouraged by the increasing awareness of drug concentration-response relationships, the mapping of drug pharmacokinetic characteristics, the advent of high-throughput computerization, and advancements in analytical technology [11]. The more recent explosion of pharmacogenetic and pharmacogenomic research has been fuelled by the tremendous amount of genetic data generated by the Human Genome Project (HGP). In 1990, the HGP began its quest to map the complete set of genetic instructions of the human genome [12,13], consisting of approximately 3.2 billion base pairs encoding up to 100,000 genes located on 23 pairs of chromosomes [14]. Although originally conceived as a 15-yr project, the HGP was essentially completed by 2001 [15]. Recent advancements in gene chip technology have ushered in a new era of gene-based medicinal and drug therapies.
PURPOSE OF THERAPEUTIC DRUG MONITORING
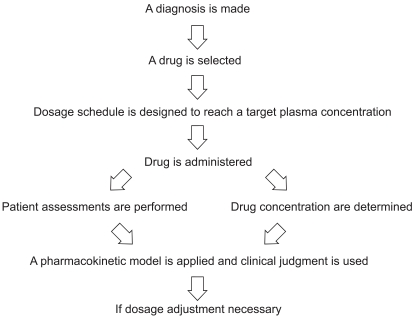
Performing TDM requires a multidisciplinary approach. Accurate and clinically meaningful drug concentrations are attainable only by complete collaboration by a TDM team, typically comprised of scientists, clinicians, nurses, and pharmacists. Excellent communication among team members is necessary to ensure that best practices in TDM are achieved (Fig. 1) [16,17].
The indications for drug monitoring have widened to include efficacy, compliance, drug-drug interactions, toxicity avoidance, and therapy cessation monitoring [18, 19] (Table 1). Plasma drug concentration measurements alone may be helpful in several circumstances, although each indication may not apply equally to every drug Measuring plasma concentrations may be helpful, however, as a low measurement reflects either poor recent compliance or undertreatment. Poor compliance is implicated if the patient is prescribed a dose that is unlikely to be associated with a measured low concentration or if a previous measurement suggested that the plasma concentration should be higher for the given dose. When initiating drug therapy, the physician may find it useful to measure the plasma drug concentration and tailor the dosage to the individual. This directive applies to all drugs, although it is most important for those with narrow therapeutic ranges such as lithium, cyclosporine, and aminoglycoside antibiotics.
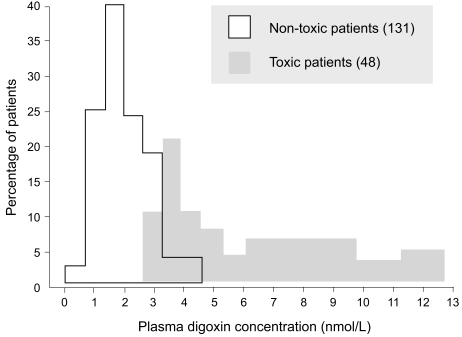
If the dosage regimen must be altered for any reason at a later stage of treatment, for example, in patients with renal failure, measuring plasma concentrations again may be helpful. Undertreatment of an established condition may be concluded if a poor clinical response is observed. However, when the drug is being used as prophylaxis, it is impossible to monitor a response. Thus, the physician can select a dosage that will produce a certain target plasma concentration. This dictum applies particularly to lithium in preventing manic-depressive attacks, to phenytoin in preventing fits after neurosurgery or trauma, and to cyclosporine in preventing transplant rejection. In all cases, plasma concentration measurements obtained and scrutinized during the early treatment stages enable the physician to avoid toxic plasma concentrations. In many cases, drug toxicity can be diagnosed clinically. For example, it is relatively easy to recognize acute phenytoin toxicity, and measuring the plasma concentration may not be necessary for diagnosis, although it may be helpful in adjusting the dosage subsequently. On the other hand, digoxin toxicity may mimic certain symptoms of heart disease, and measuring the plasma concentration in cases in which toxicity is suspected may be helpful in confirming the diagnosis. In a study by Aronson and Hardman [20], measurement of the plasma digoxin concentration in 260 patients treated with digitalis lanata preparations (digoxin, lanatoside C, betamethyl-digoxin) enabled the monitoring of certain outcomes that would not be apparent otherwise. Notably, the important overlap between "toxic" and "nontoxic" plasma concentration values limits use of the method in the diagnosis of digitalis toxicity (Fig. 2) [20]. However, in digitalis-treated patients with toxicity associated with digitalis plasma concentrations under 2.0 ng/mL, the method can detect digitalis sen-sitivity. Aronson and Hardman [20] determined that a dosage selection based on plasma drug concentration assessment led to a decrease of digitalis toxicity to below 4%. This method is not yet widely available. Thus, it should be noted that plasma digoxin concentration measurements should be obtained and evaluated in digitalis-treated patients with borderline renal function, in aged subjects, and in patients with rapid atrial fibrillation who require higher digitalis doses for heart rate control (Fig. 3) [21].
Similarly, nephrotoxicity of aminoglycoside antibiotics is difficult to distinguish clinically from that caused by a severe generalized infection. Thus, measuring aminoglycoside plasma concentrations may help to distinguish between toxicity and infection. If the potential for a drug interaction is suspected, then measurement of the plasma concentration may guide subsequent changes in dosage. For example, when giving a thiazide diuretic to a patient taking lithium, measuring the plasma lithium concentrations is helpful to avoid toxicity. When the patient's renal function remained stable, and he developed no signs or symptoms of digoxin toxicity. To our knowledge, no case reports have associated significant fluctuations of digoxin plasma concentrations corresponding to the timing of oral amiodarone administration. However, clinicians should be aware that digoxin plasma concen-trations may not correlate with digoxin tissue concentrations in this setting. When a loading dose of oral amiodarone is required in a patient receiving digoxin, the digoxin dosage should first be reduced, and digoxin therapy should be adjusted based on any signs and symptoms of digoxin toxicity [22]. This approach also applies to theophylline when erythromycin is added to the regimen. Conversely, measuring the whole blood cyclosporine concentration will help to avoid undertreatment if rifampicin is added.
MEASURING PLASMA DRUG CONCENTRATION IN THERAPEUTIC DRUG MONITORING
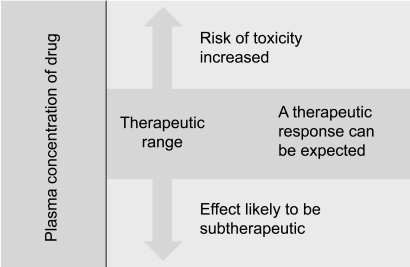
The contribution of pharmacokinetic variability to differences in dose requirements can be identified by measuring the drug concentration at steady state and modifying the dose to attain a desired concentration known to be associated with efficacy. However, there is substantial inter-individual pharmacodynamic variability at a given plasma concentration [23], hence a range of concentrations rather than a single level is usually targeted. For a limited number of drugs for which there is a better relationship between plasma or blood concentration-response than dose-response, the measurement of plasma or blood concentrations has become a valuable surrogate index of drug exposure in the body [16].
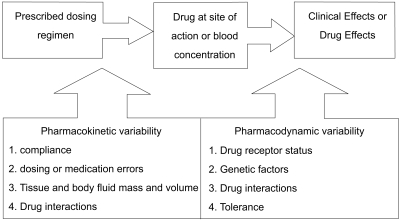
Pressures continue within the health care system to provide services at the lowest possible cost. Thus, the role of many drug assay laboratories is to measure the concentration of a therapeutic drug in a blood sample and relate this number to a therapeutic range published in the literature. Therapeutic drug measuring is only one part of TDM that provides expert clinical interpretation of drug concentration as well as evaluation based on pharmacokinetic principles. Expert interpretation of a drug concentration measurement is essential to ensure full clinical benefit. Clinicians routinely monitor drug pharmacodynamics by directly measuring the physiological indices of therapeutic responses, such as lipid concentrations, blood glucose, blood pressure, and clotting. For many drugs, either no measure of effect is readily available, or the method is insufficiently sensitive [24]. Therefore, the process of TDM is predicted on the assumption that a definable relationship exists between dose and plasma or blood drug concentration, and between the blood drug concentration and pharmacodynamic effects (Fig. 4) [16]. Measuring the plasma drug concentration may guide clinicians to stop treatment under two known circumstances. First, treatment should cease if the plasma digoxin concentration is below the therapeutic range in a patient whose clinical condition is satisfactory so that digoxin withdrawal is unlikely to lead to clinical deterioration. Note that this use of the plasma concentration measurement depends on the concept that there is a lower end to the therapeutic range. This is not true for other drugs, particularly phenytoin. If there is no response to lithium and the serum concentration is at the upper end of the therapeutic range, then increased dosage is unlikely to be beneficial, and the risk of toxicity is high. Withdrawal of lithium and the use of a different treatment would be justified. Drug concentration measurements are requested to assist the management of a patient's current medication regimen or to screen for a medicine. Procedures may also be implemented to assess whether requests for drug assays are warranted before the assays are actually performed, thereby ensuring the rational utilization of resources. This is often time consuming for senior personnel, but can be cost-effective as it may prevent expensive tests that do not assist either immediate or long-term patient management [16].
For a small number of drugs, measuring the plasma concentration is helpful in clinical practice. Table 2 presents the criteria that must be satisfied for the drug plasma concentration to be useful [19].
Even for drugs that fulfill these criteria, some controversy exists about the usefulness of monitoring their plasma concentrations [20]. First, it has been argued that no good evidence demonstrates that targeting plasma concentrations improves the therapeutic outcome [24, 25], and that the therapeutic value of plasma monitoring must be tested [26]. However, these arguments ignore the underlying principle: a stronger relationship exists between plasma concentration and effect than between dose and effect [16], suggesting that it should be possible to improve therapy with a drug by monitoring its plasma concentrations. Second, it is argued that the value of the technique is reduced by problems in defining therapeutic ranges, such as those encountered when conditions alter a drug's pharmacodynamic effects [25,26]. However, this argument merely emphasizes the need for proper interpretation of plasma drug concentrations under such conditions [19]. Third, some argue that the plasma concentration itself is being treated rather than the patient [27], and that monitoring is rendered useless by, for example, an inappropriate timing of sampling [26]. We argue that this last point indicates that the information provided by plasma drug concentration monitoring is being misused [19]. There is no justification for routine measurements of plasma drug concentrations without a definite purpose. Indeed, routine measurement of the plasma drug concentration without a clear purpose is as irresponsible as obtaining no measurement at all.
ANALYTICAL ISSUES IN THERAPEUTIC DRUG MONITORING
As stated previously, the practice of therapeutic drug monitoring requires the orchestration of several disciplines, including pharmacokinetics, pharmacodynamics, and laboratory analysis. The analytical impact on determining pharmacokinetic parameters is not well appreciated. Analytical goals in therapeutic drug monitoring should be established by determining the nature of the problem to be solved, selecting the appropriate matrix and methodology to solve the problem, and developing valid analytical schemes that are performed competently with appropriate quality and interpreted within the framework of the problem [28].
If plasma drug concentration measurements are to be of any value, attention must be paid to the timing of blood sampling, the type of blood sample, the measurement technique, and the interpretation of results. First, it is vital to obtain the blood sample for measuring the drug concentration at the correct time after dosing. Errors in the timing of sampling are likely responsible for the greatest number of errors in interpreting the results. For most drugs, the blood sample can be drawn into a heparinized tube or allowed to clot, and there are no important restrictions on storage before measurement. For lithium and aminoglycosides, however, the blood samples should be allowed to clot, and should be separated within 1 h. For cyclosporine, it is important to consult the local laboratory for details on the proper sampling technique and post-dosage timing. The laboratory must ensure that the assay used is as reliable and specific as possible and that appropriate quality control is undertaken. Method validation is becoming a more universally important consideration. The pharmaceutical industry has mounted a worldwide effort to harmonize the concepts used in validation, which are summarized in Table 3 [29]. Ensuring the accuracy and specificity of assays used by the clinical laboratory to measure serum drug concentration is critical. Historically, drug testing laboratories developed their assay procedures using a variety of analytical methods ranging from radioimmunoassay to high-performance liquid chromatography (HPLC) procedures. Currently however, the vast majority of drug assays performed in the clinical setting are some variant of commercially available immunobinding assay procedures [30]. The most commonly used procedures are fluorescence polarization immunoassay (FPIA), enzyme immunoassay (EMIT), and enzyme-linked immunosorbant assay (ELISA) [31,32]. These assays are specific; however, in certain cases, metabolites or other drug-like substances are also recognized by the experimental antibody [33-35]. Most such assay interferences are the result of cross-reactivity with the drug's metabolites, but in some cases, endogenous compounds or drugs with similar structures can cross-react, resulting in either a falsely elevated or decreased assayed drug concentration reading [35-39].
PRACTICAL ISSUES IN THERAPEUTIC DRUG MONITORING
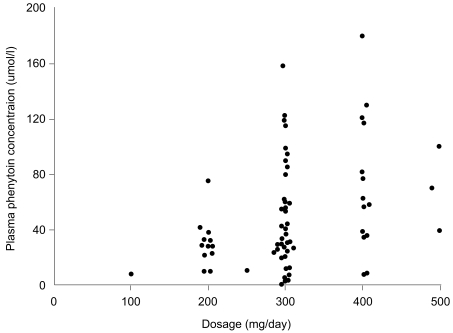
Ideally, a quality drug assay should be performed within a time frame that is clinically useful. In large chemical pathology laboratories staffed by highly skilled scientists and equipped with state-of-the art automated analyzers, many clinicians assume that the results will be accurate. Therefore, analytical laboratories should ensure that procedures are in place to obtain any missing information from the drug assay request that may be needed for appropriate clinical interpretation of the results, such as dosage regimen, time of blood sampling, and that the accuracy, precision, sensitivity, and specificity of each assay is documented and assessed regularly. Wherever possible, the assay performance should be evaluated using an external quality assurance program that provides a rapid turn-around time for results and comprehensive feedback on the assay performance, and that has a large number of subscribers. The assay results should be available quickly, preferably within 24 h of receiving the sample, as the most important uses of the measurements are during dosage adjustments and in diagnosing toxicity, when rapid decisions must be made. Indeed, there is evidence that on-site measurement of antiepileptic drugs has an immediate impact on clinical decision making processes and outcomes [40]. The most important consideration in interpreting the plasma drug concentration is tailoring the treatment to the patient's physiological needs. In doing so, the clinician should take into account not only the concentration but also other clinical features that may affect the relationship between concentration and clinical effects. Thus, it is important for the clinician to know how to interpret the plasma concentration results in the context of the patient's condition, rather than making a predetermined guess as to what that measurement might mean [19]. The information needed to interpret a drug concentration result is given in Table 2. Patient demographic characteristics are critically important so that the contributions of age, disease state, ethnicity, and other variables to inter-individual variation in pharmacokinetics and pharmacodynamics can be considered. The clinician presenting a drug assay request must communicate these details effectively to the members of the TDM team. Once the decision to monitor the concentration of a therapeutic drug has been made, it is important that a biological sample is collected to provide a clinically meaningful measurement. An appropriate pharmacokinetic evaluation requires the acquisition of properly timed blood specimens [41]. To interpret a blood plasma concentration properly, the TDM team must be informed as to when a plasma sample was obtained in relation to the last dose administered and when the drug regimen was initiated. If a plasma sample is obtained before distribution of the drug into tissue is complete, for example with digoxin, the plasma concentration will be higher than predicted on the basis of dose and response. Peak plasma concentrations are helpful in evaluating the dose of antibiotics used to treat severe, life-threatening infections. Although serum concentrations for many drugs peak 1 to 2 h after an oral dose is administered, factors such as slow or delayed absorption can significantly delay the time at which peak serum concentrations are attained. Therefore, with few exceptions, plasma samples should be drawn at trough or just before the next dose (Css min; minimal steady state concentration) when determining routine drug plasma concentrations. These trough levels are less likely to be influenced by absorption and distribution problems [42]. If a patient is administered a drug repeatedly, the drug and its metabolites will accumulate in the body. Eventually, when the amount being given is equal to the amount being eliminated, an equilibrium or "steady state" is reached. The time required to reach this steady state depends only on the half-life of the drug. After 5 half-lives, over 95% of a drug will have accumulated, and for practical purposes, steady state has been achieved. The plasma concentration can be measured before this steady state has been reached, but the timing of the sample must be considered when interpreting the results. Blood samples should be collected once the drug concentrations have attained steady state, for example, after at least 5 half-lives at the current dosage regimen. Levels approximating steady state may be reached earlier if a loading dose has been administered. However, drugs with long half-lives should be monitored before steady state is achieved to ensure that individuals with impaired metabolism or renal excretion are not at risk of developing toxicity at the initial dosage regimen prescribed, as can occur with amiodarone and perhexiline. If drug toxicity is suspected, then the plasma concentrations should be monitored as soon as possible. Likewise, an immediate assay might be indicated in cases of poor therapeutic control, as in rapid atrial fibrillation, when loading doses could be useful. To interpret the result, details of the dosage regimen (dose and duration) are essential. Blood or plasma concentrations change throughout a dosage interval, and the time of the blood sample draw relative to the time of dose administration must be known to enable sensible interpretation. Absorption is variable after oral administration, and blood samples should be collected in the elimination phase rather than in the absorption or distribution phases. Usually blood samples are collected at the end of the dosage interval (trough level). For antibiotics administered intravenously, peak concentrations are also measured at 30 min following infusion cessation. For aminoglycoside antibiotics, both peak and trough concentrations are important measurements. If the drug has been administered by bolus injection, samples should be taken at least 1 h post-dosage to avoid overlapping the distribution phase. Concentrations measured at these time points can be compared with published therapeutic ranges, which are usually based on prospective studies that relate trough drug concentrations measured at steady state to pharmacodynamic responses. If a given dose of a drug produced the same plasma concentration in all patients, there would be no need to measure the plasma concentration of the drug. However, people vary considerably in the extent to which they absorb, distribute, and eliminate drugs. Ten-fold or even greater differences in steady-state plasma concentrations have been found among patients treated with the same dose of important drugs such as phenytoin, warfarin, and digoxin (Fig. 5). These differences are due in large part to differences in drug formulations, patient genetic variation, underlying disease, environmental effects, and drug-drug interactions. Therefore, measuring the plasma concentration of a drug allows the doctor to track the dosage to the individual patient and to obtain the maximum therapeutic effect with minimal risk of toxicity. Information about plasma concentration is helpful for a number of drugs in clinical practice. Several criteria must be satisfied for the plasma concentration of a drug to be useful. If it is easy to measure the therapeutic or toxic effects of a drug directly, the plasma drug concentration gives little additional information about drug action. On the other hand, if it is difficult to measure the therapeutic effects of a drug, then measuring the plasma concentration helps to tailor the dose within the appropriate therapeutic range. There is little point in measuring the plasma drug concentration if it will not give interpretable information about the therapeutic or toxic state of the patient; for example, if there is a subtherapeutic concentration of digoxin in a patient with compensated heart failure and sinus rhythm, digoxin may be withdrawn without fear that the patient's heart failure will worsen. Additional criteria include a low toxic-to-therapeutic ratio and the presence of active metabolites. Even if a drug satisfies these criteria, interpretation of the plasma drug concentration may be rendered difficult by the presence of a metabolite with a distinct therapeutic or toxic activity. If active metabolites are produced, both the parent drug and the metabolites must be measured to provide a comprehensive picture of the relationship between the total plasma concentration of the active compounds and the clinical effect. This is usually not possible in routine monitoring, which limits the usefulness of plasma concentration measurements of, for example, procainamide, which is metabolized to N-acetylprocainamide (acecainide), which has equipotent antiarrhythmic activity. Drug interactions, electrolyte balance, acid-base balance, age, bacterial resistance, and protein binding are some factors that modify the effect of the parent drug for a given drug plasma concentration if total drug concentration is measured.
PHARMACOECONOMIC IMPACT OF THERAPEUTIC DRUG MONITORING
Over the last 30 years, in response to the lessons learned from using TDM and growing concerns among clinicians and the public about rising health care costs, the principles of pharmacoeconomics are now being applied to various fields, including TDM [43]. As an intervention method, TDM purports to improve patient responses to important life-sustaining drugs and to decrease adverse drug reactions. Furthermore, the resources consumed by TDM methods will likely be regained by positive outcomes, including decreased hospitalizations, and thus TDM is an appropriate candidate for an economic outcomes evaluation [44,45]. Donabedian's proposal [46] advocates the structure-process-outcome method for assessing the quality of health care practices. His evaluation of the structure component in this method includes factors related to the construction of a health care delivery system, including its buildings, equipment, staff, and patient mix; the process component includes the activities involved in health care delivery services; and the outcome component examines the effect of a health care intervention on patient outcome, as well as the impact of the economic performance of the health care system [46]. Extending Donabedian's analysis to TDM, with of structural components include the TDM testing equipment and facilities, qualifications of the clinical and laboratory staff, the presence of a TDM service, monitoring supervision, and administrative organization. The process component involves procedures such as assuring appropriate indications for ordering serum drug levels, timing of sample collections, communication of results to the clinician, and monitoring for appropriate clinician responses to treatment recommendations and for patient response to treatment. Finally, the outcome measures to assess the TDM effectiveness include assessing the incidence of drug-induced adverse reactions, cure rates, mortality rates, and cost savings associated with a TDM service [47]. A pharmcoeconomic analysis of the impact of TDM in adult patients with generalized tonic-clonic epilepsy showed that patients undergoing TDM had much more effective seizure control, fewer adverse events, better earning capacity, lower costs to the patient, savings from lower hospitalizations per seizure, and greater chances of remission [48]. A meta-analysis of TDM studies, albeit on a limited number of drugs, showed that TDM does appear to be beneficial for patients taking theophylline or digoxin [49]. The same group also concluded that a clinical pharmacokinetic service run by clinical pharmacists had a significant influence on the proportion of patients with desirable serum drug concentrations. Furthermore, the service reduced the proportion of inappropriately collected samples. TDM of aminoglycosids is an important approach to reduce the incidence of aminoglycosid toxicity while maximizing efficacy parameters, such as optimizing the peak-to-minimal inhibitory concentration ratio. Several patient-oriented studies have reported high cost-effectiveness of dose individualization using TDM [50-53]. Although vancomycin is considered to be less nephrotoxic than the aminoglycoside, a relationship seems to exist between serum concentrations and toxicity and efficacy [54]. All of the current immunosuppressants exhibit large inter- and intra-individual variability in pharmacokinetic factors, and in several concentration-controlled trials, it has been demonstrated that blood concentration is a better predictor of clinical efficacy than dose [55]. Over the first decade, many consensus documents have been published that address the need for and methodology of immunosuppressive drug monitoring, with the most recent publication including important guidelines and recommendations for cyclosporine, silorimus, and tacrolimus adminiatration [56]. With the exception of aminoglycoside, however, there remains a dearth of well-designed studies investigating the added value and cost-effectiveness of TDM. For therapy with antiepileptic drugs, digoxin, psychiatrics, and immunosuppressant drugs, TDM is considered as the standard of care despite the lack of formal cost-effectiveness data [1].
SUMMARY
The use of TDM requires a combined approach encompassing pharmaceutical, pharmacokinetic, and pharmacodynamic techniques and analyses. The appropriate use of TDM requires more than a simple measurement of patient blood drug concentration and a comparison to a target range. Rather, TDM plays an important role in the development of safe and effective therapeutic medications and individualization of these medications. Additionally, TDM can help to identify problems with medication compliance among noncompliant patient cases. When interpreting drug concentration measurements, factors that need to be considered include the sampling time in relation to the dose, the dosage history, the patient's response, and the desired clinical targets. This information can be used to identify the most appropriate dosage regimen to achieve the optimal response with minimal toxicity [57,58].



 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print





