


INTRODUCTION
Parental genomic imprinting is the phenomenon in which the differential behavior of a gene depends on the sex of the transmitting parents. Either an imbalance of parental chromosomes in the embryo or an aberrant imprinting of these genes is implicated in an increasing number of genetic disorders1–7).
Type 1 diabetes mellitus is caused by the autoimmune destruction of pancreatic β-cells8). It is a polygenic disorder, linked to several genetic loci9, 10) and therefore, phenotype expression is influenced by a number of susceptibility genes as well as environmental factors. Recent genome wide linkage studies indicate that genes in the HLA class II region provide the major genetic susceptibility to IDDM11, 12). The insulin gene (INS) region on chromosome 11p15.5 (IDDM2) has also been shown to contribute to IDDM susceptibility in many populations13–18). The remaining genetic component is largely unknown, although recent studies have revealed the presence of several loci showing a significant genetic linkage12, 19). That one or more of these loci may be subject to parental imprinting was first suggested in the observation that IDDM is transmitted by diabetic fathers more than twice as often as by diabetic mothers20). Further, more direct evidence that IDDM transmission is modified by parental imprinting was generated by a large study of the IDDM2 locus in a French and mixed North American population14, 21–23). But it is controversial whether IDDM2 transmission is positively associated with type I diabetes in Asians.
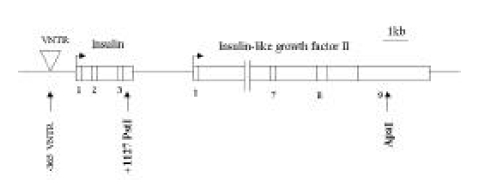
The IDDM2 locus was previously mapped to a 4.1-kb region spanning the insulin gene and a variable number of tandem repeats (VNTR) locus on the human chromosome 11p15.516). Genetic susceptibility to IDDM is encoded by several loci, one of which (IDDM2) maps to a variable number of tandem repeats (VNTR) minisatellite, upstream of the insulin gene (INS). The short class I VNTR alleles (26–63 repeats) predispose to IDDM, while class III alleles (140–210 repeats) have a dominant protective effect24). Class III alleles are associated with marginally lower INS mRNA levels than class I in the human adult and fetal pancreas in vivo. Class I alleles are found with higher frequencies in type I diabetic patients than the class III alleles14, 24). The shorter alleles of class I predispose to IDDM, while the longer class III alleles are protective. These may be related to type 1 diabetes pathogenesis, as insulin is the only known cell-specific IDDM autoantigen. INS is a target of tissue-specific VNTR transcriptional effects and is the only known cell specific autoantigen in type 1 diabetes25–27). Additionally, INS was recently shown to be imprinted in the yolk sac but not in the pancreas of mice28).
The IGF2 gene located immediately 3′ to INS is maternally imprinted in mice and in humans29, 30). This favors IGF2 as the gene involved in IDDM transmission. However, in human peripheral lymphocytes, in which lectin stimulation induces IGF2 transcription, imprinting is relaxed and the maternal copy is seen in most but not all individuals31, 32). This variable relaxation of imprinting may be the genotype-dependent trait postulated by Bain et al.15) which explains either the presence or absence of imprinting phenomena in different populations. Functionally, IGF2 is also considered a candidate for the IDDM2 effect due to its well-characterized role as a mitogenic peptide with important autocrine/paracrine effects on the thymus, lymphocytes and pancreas33, 34).
In this report, we studied patterns of IDDM2 parental transmission and the imprinting effects of the INS and IGF2 genes in Korean type I diabetic patients.
MATERIALS AND METHODS
Samples Whole blood samples were obtained from patients diagnosed with type 1 diabetes mellitus by the criteria of the National Diabetes Data Group. The samples used in the genotype and allele-specific expression analyses were obtained from 26 patients with no family history of diabetes and 16 patients with a family history of diabetes. The patients (aged 6–16) and their parents were recruited through the pediatric department of Dongsan hospital and the Diabetic Camp of the Daegu and Kyungbook.
Nucleic Acid Preparation
Genomic DNA extraction. Mononuclear cells were isolated from 10 mL of whole blood samples using ammonium chloride erythrocyte lysis buffer. Genomic DNA was purified using 100 μg/μL proteinase K and 0.5% sodium dodecyl sulfate and then extracted in phenol/chloroform/isoamylalcohol (25:24:1).
Total RNA extraction and cDNA synthesis. Mononuclear cells were isolated from 10 mL of whole blood samples using Ficoll-Paque solution (Pharmacia Biotech., USA). Total cellular RNA was prepared as RNA Zol B (TEL-Test. INC., USA). To generate cDNA, 2 μg total RNA was reverse-transcribed in 20 μg of reaction mixture containing 5 mmol MgCl2, 1 mmol dNTP, 1 U/μL RNase inhibitor, 2.5 U/μL MuLV reverse transcriptase and 2.5 M oligo d (T)16 for 1h at 42°C.
Polymerase chain reaction (PCR). A set of primers was generated to PCR-amplify a nucleotide segment, including the PstI+1127 restriction fragment length polymorphism (RFLP), in the 3′ untranslated region of INS24). Sequences of the primer pair were as follows (5′–3′): INPsA, TGGTGCAGGCAGCCTGC; INPsB, GGTGGTTCAAGGGCTTTATT. Likewise, a primer set used to amplify IGF2-exon 9, including an Apa I RFLP, was made as follows (5′–3′): IGApA, CCTGGACTTTGAGTCAAATTGG; IGApB, CCTCCTTTGGTCTTACTGGG30). 500 ng genomic DNA and 4 μg RT-product were subjected to PCR for 35 cycles in a mixture containing each primer pair exposed to the following conditions: for INS PCR, initial denaturation at 95°C for 10 min by 35 cycle of 92°C for 1 min, 55°C for 1 min and 72°C for 2 min.
RFLP Analysis. After PCR products were purified using the wizard PCR preps DNA purification system (Promega, USA), they were digested with 30 U of PstI or ApaI at 37°C overnight and electrophoresed on 2% agarose gel or 6% polyacrylamide gel. The gel was then stained with ethidium bromide for visualization.
Expression studies. Expression of the INS and IGF2 genes was assessed using the same PCR primers. GAPDH served as an internal control for the reaction. A primer set to amplify GAPDH was made as follows (5′–3′): S, CGTCTTCACCACCATGGAGA; AS, CGGCCATCACGCCACAGTT35). PCR amplification was carried out under the following conditions: initial denaturation at 95°C for 10 min by 30 cycles of 94°C for 30 sec, 60°C for 30 sec and 72°C for 30 sec. PCR products were electrophoresed on 2% agarose gel followed by ethidium bromide staining. Samples were quantified by densitometry using a Molecular Analyst version 1.4 image analyzer (Gel Doc 1000, Bio-RAD, USA).
RESULTS
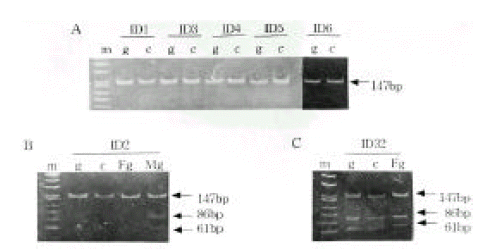
Genotype Determination. The families of type I diabetic patients were studied. Sixteen of 42 families were found to include type I diabetic members, and 3 of these diabetic patients had a diabetic mother. None of the patients had a diabetic father. A total of 42 type 1 diabetic patients were genotyped for the PstI+1127 and ApaI polymorphism in INS and IGF2, respectively (Table 1). The absence of a restriction site in INS is indicated as the ‘+’ allele, while the allele containing the cutting site is termed ‘−’ allele. The absence and presence of the restriction site in IGF2 is indicated as the ‘−’ and ‘+’ allele, respectively. The INS PCR product was 191 bp long, although there was more than one PstI restriction site present (Figure 1). Digestion of the PCR products with PstI produced, in the presence of two restriction sites, fragments of size 86 bp and 61 bp. In the absence of restriction sites, a fragment of 147 bp was generated. The IGF2 PCR product was 236bp long. Digestion of the PCR products with ApaI, in the presence of a restriction site, produced a 173 bp fragment and, in the absence of restriction sites, a fragment of 236 bp was generated. PstI digestion of PCR products from genomic DNA demonstrated that 36 (97.3%) of the 37 IDDM patients tested were homozygous for the PstI (+/+) allele (Table 1, Figure 2A). PCR of genomic DNA revealed that 29 (72.5%) of the 40 type 1 diabetic patients were heterozygous (ApaI (+/−)), while the others were homozygous (ApaI (−/−)) for the ApaI RFLP in IGF2 (Table 1). The frequencies of the INS/PstI (+/+) homozygous genotype were found to be much higher than the INS/PstI (+/−). heterozygous genotype. These results confirm that the INS/PstI (+/+) homozygote is the dominant genotype in Korean type I diabetic patients.
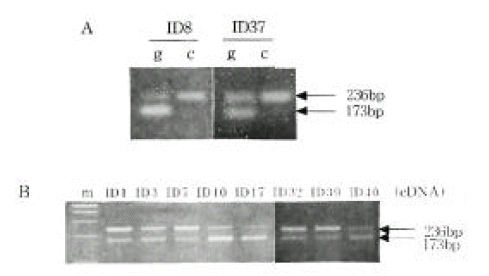
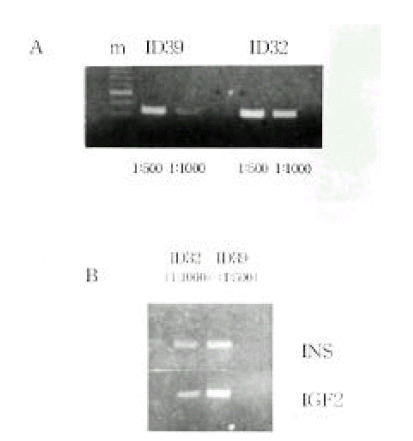
Allele-specific expression of INS and IGF2. We examined the presence of each allele in the RNA transcript from lymphocytes identified as heterozygous, PstI (+/−) and ApaI (+/−). In Figure 2B and 2C, the amplification-digestion products of INS genomic and complimentary DNA of two families are shown. One of the families expressed heterozygous and all were found to be both alleles for the PstI polymorphism in their genomic DNA (Figure 2C). In the other family, the mother was PstI (+/−) heterozygous and the patient was PstI (+/+) homozygous (Figure 2B). We could not demonstrate that the INS genotype was associated with parental imprinting in IDDM transmission because most of the patients had PstI (+/+) INS genotypes. As illustrated in Table 2, both IGF2 alleles resulted in ApaI digestion in 21 of the 24 ApaI (+/−) heterozygotes. In Figure 3B, a series of individuals are shown who are characterized by biallelic IGF2 expression. In Figure 3A, 2 individuals who exhibit monoallelic IGF2 expression are listed. Therefore, the presence of both of these alleles in RNA indicates that IGF2 imprinting is at least partially relaxed in these cells. The relative intensities of the PstI (+/−) and PstI (+/+) RNA transcripts were measured using semi-quantitative RT-PCR. We found a 3-fold higher level of RNA expression from the Pst I (+/−) transcript compared to that of the Pst I (+/+) transcript in peripheral leukocytes (Figure 4). We also measured the relative intensities of IGF2 RNA transcripts in the same type 1 diabetic patients. In this case, the expression of the Pst I (+/−) transcript was greater than that of the Pst I (+/+) transcript, although these patients showed biallelic IGF2 expression (Figure 4).
DISCUSSION
Warram et al20) proposed that IDDM is transmitted to offspring by diabetic fathers more than twice as often as by diabetic mothers. This is the first indication that IDDM is related to parental imprinting. Following studies went on to examine the evidence of a linkage between IDDM transmission and parental imprinting14, 21–24, 30, 32). Of the families included in this study, 16 out of 42 were found to include type I diabetic members. Of these, 3 had a diabetic mother, although none had a diabetic father. These findings contrast with those from a previous report20). We cannot explain the relationship between IDDM transmission and parental imprinting. INS and IGF2 are considered prime candidate genes for the IDDM2 locus and for IDDM2-encoded susceptibility14, 21, 22, 24). This is especially true since IGF2 is exclusively expressed by the paternal copy in most tissues33), which leads to our hypothesis that IDDM-encoded susceptibility is mediated by IGF232). To test this, we examined genotype determination and allele-specific expression using mononuclear cells from IDDM patients and their parents. Twenty-nine of 40 type 1 diabetic patients express heterozygous and the data of parental genotype were available in only 9 of 29 type 1 diabetes patients. Only 3 cases of IGF2 mRNA revealed maternal origin. There seems to be polymorphic relaxation of IGF2 imprinting in leukocytes. But we need to require a much larger number of samples for statistical validity. However, these results are similar to the findings of previous reports that showed that IGF2 imprinting is relaxed in human peripheral lymphocytes36, 37). This relaxation was incomplete as maternal allele mRNA was systematically at a lower level than paternal. The paternal/maternal ratio varied widely among individual subjects. Variable relaxation of IGF2 imprinting seen in human leucocytes is not dependent on the presence of a class I versus a class III VNTR36, 37). Conclusively, mRNA from both IGF2 alleles was seen and it indicated relaxation of parental imprinting of IGF2 in leukocyte samples. Our results were not enough to evaluate the IGF2 imprinting effect but we could not exclude IGF2 as the candidate gene involved in IDDM2. However, the absence of an IGF2 imprinting effect suggests that, if there is a parental component in IDDM transmission, it is not mediated through IGF2. INS was shown to be imprinted in the yolk sac but not in the pancreas of mice28). Maternal imprinting of the INS gene has originally been considered the most favorable explanation for the observed gender-related difference. However, previous studies demonstrated a biallelic expression of INS in pancreatic tissues with monoallelic and biallelic expression in the thymus of human fetuses. This suggests that INS is probably not imprinted in these tissues and is expressed tissue-specific pattern36–40).
We studied the specific pattern of INS genotypes and the association of their allele-specific expression and INS imprinting in peripheral mononuclear cells from Korean type I diabetic patients. Thirty-six of the 37 IDDM patients were found to be PstI (+/+) and in 1 case, PstI (+/−). We could not demonstrate that the INS genotype was associated with parental imprinting in IDDM transmission because the majority of Koreans had PstI (+/+) INS genotypes. Also, there was no information about parental imprinting pattern of INS genotype in Asia because it had been difficult to get examples due to the same reason. This is the limitation to the study of INS genotype transmission in Korea. However, we can not exclude genetic and environmental differences between the patients of Korea and western countries, in which the occurrence of type 1 diabetes is found to be either rare or more common, respectively.
The RT-PCR assay of Bennett et al24). provides some evidence for a quantitatively different expression of the INS ‘+’ and ‘−’ forms in the two heterozygotes. Bennett et al demonstrated 1.5 to 3-fold more expression of class I than class III haplotypes. We also measured the relationship between the genotypes and INS transcription levels using semi-quantitative RT-PCR. Our findings were similar to Bennett et al.24) in that we found a 3-fold higher level of expression from PstI (+/−) with class I/III compared to PstI (+/+) with class III/III. However, Pugliese et al.40) observed that class III VNTR/PstI (−) mRNAs were transcribed at higher levels (2.5-fold on average) than class I VNTR/PstI (+) mRNA in the thymus and inverse correlation was found in pancreatic islet cell samples. VNTR alleles correlate with differential INS mRNA expression in the thymus where protective class III VNTRs are associated with higher steady-state levels of INS mRNA expression. This finding could explain the dominant protective effect of class III VNTRs because the elevated level of proinsulin in the thymus enhances negative deletion of insulin specific T-lymphocytes which play a critical role in the pathogenesis of type 1 diabetes14, 16, 24). Our results are consistent with those of previous reports and indicate a different RNA transcriptional level for INS genotype according to VNTR class. However, our studies of the relationship between INS imprinting and allele-specific expression need to be conducted in a larger population of type I diabetic patients. Further study of both the genetic behavior of specific alleles within VNTR classes and their effect on transcription levels will be needed to conclusively confirm and define the association between INS transcription levels and IDDM parental transmission. Then, we may be able to better confirm parental genomic imprinting of IDDM2 transmission in the Korean type I diabetic population.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print





