


INTRODUCTION
Chronic liver disease is one of the most frequent causes of bleeding, which can manifest itself as epistaxis, gum bleeding, ecchymosis, and gastrointestinal hemorrhaging. The most significant bleeding problem among patients with chronic liver disease is blood loss due to portal hypertension. Nearly 70% of the upper gastrointestinal bleeds in cirrhotics, are caused by the rupture of esophageal varices1). Other less frequent causes of bleeding include, congestive gastropathy, gastric varix, peptic ulcer diseases, and gastritis. Only on rare occasions has the non-gastroesophageal vein been the cause of bleeding2-7). One case of spontaneous rupture of the lumbar artery in a patient with alcoholic liver cirrhosis was reported8); however, spontaneous bleeding of the lateral thoracic artery in a patient with liver cirrhosis has not been reported. Here, we report a case of spontaneous rupture of the lateral thoracic artery in patients with liver cirrhosis and coagulopathy.
CASE REPORT
A 47-year-old male patient who was a smoker (10 pack-year) and a former alcoholic drinker who had quit after being diagnosed with cirrhosis of the liver. The patient also experienced a change in mental state. In addition, this particular patient was also diagnosed with abdominal distension, dyspnea on exertion, and lower extremity swelling. In addition, the patient had also been a diabetic for four years as well as having been diagnosed with alcoholic liver cirrhosis a year and a half prior to his death. The patient also had a lapse medical surveillance for a year and a half and was without medication. His primary complaints developed about one month before his death. Upon physical examination, the patient's vital signs were stable, although he appeared pale and mildly dyspneic with cold extremities as well as yellowish skin discoloration and icteric sclera. We also noted an audible decrease in breathing sound on left lower lung field. The abdomen was protuberant distended with tense fluid, in addition to a decreased bowel sound. Despite this, there were no tender areas or rebound tenderness. Both lower extremities had pretibial pitting edema, palmar erythemas, and the presence of spider angiomas.
The laboratory data on the patient were: white blood cell count (WBC), 6,670/mm3; hemoglobin, 4.5 g/dL; platelet count, 163,000/mm3; prothrombin time, 23.8 sec (INR, 2.20); partial thromboplastin time, 58.8 sec; fibrinogen, 0.99 g/dL; glucose, 58 mg/dL; albumin, 1.97 g/dL; AST/ALT, 52/20 IU/L; total bilirubin, 2.77 mg/dL; alkaline phosphatase, 98 IU/L; gamma glutamyl transferase, 24 IU/L; sodium, 135 mmol/L; and potassium, 5.01 mmol/L. A paracentesis performed on the patient revealed the following results: WBC, 55/mm3; albumin, 0.23 g/dL; and glucose, 70 mg/dL. The patient was transferred on the 11th hospital day to our department after management of blood sugar level in the department of endocrinology.
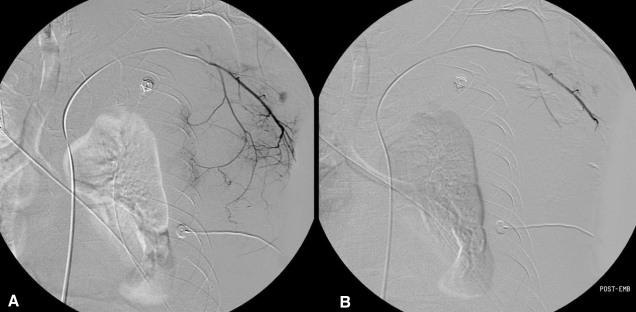
A chest PA revealed a left pleural effusion. On the 12th hospital day, the patient complained of a sudden left chest wall pain as well as swelling, and his vital signs were the following: blood pressure, 140/90 mmHg; heart rate 92, beats/min; respiratory rate, 20/min; and body temperature, 36.4Ōäā. The patient's hemoglobin was 3.9 g/dL and the chest CT revealed a 13├Ś7 cm hematoma in the left chest wall (Figure 1). The patient was immediately treated with packed RBC's, an fresh frozen plasmas transfusion, and an emergency angiography which revealed active bleeding at the branch of left lateral thoracic artery. Thus, an embolization using gelfoam was performed (Figure 2). After the procedure, the patient's vital signs were stabilized. One day after the first embolization, the patient once again complained of left chest wall swelling and pain. A repeated chest CT showed a much increased hematoma at the same site (Figure 3), and a second angiography relealed active bleeding from all the branches of left lateral thoracic artery as well as some lower intercostal arteries. As a result, a repeated embolization with a contour particle was performed (Figure 4).
On the 19th hospital day, the following patient data was recorded: blood pressure, 100/60 mmHg; pulse rate, 100 beats/min; and urine output <10 mL/hour. Despite the transfusion, the patient's hemoglobin level was 7.2 g/dL, BUN/ creatinine was 95.5/3.47 mg/dL, prothrombin time was 22.6 seconds (INR 2.06), and partial thromboplastin time was 53.9 seconds. We recommended repeated embolization, but the patient's family refused. The patient expired on the 19th hospital day despite best supportive care.
DISCUSSION
Liver disease is a frequent cause of hemostatic abnormalities, which may lead to overt or occult bleeding. Clinical manifestations of hepatic coagulopathy include upper and lower gastrointestinal hemorrhaging, easy bruising, and bleeding from gums, nose or the female genital tract. Bleeding from the gastroesophageal varix in combination with extensive collateral circulation is the most common and significant problem in patients afflicted with cirrhosis of the liver. About 30% of subjects with esophageal or gastric varices resulting from cirrhosis have an episode of gastrointestinal bleeding in their lifetime9).
Coagulopathy in combination with cirrhosis are manifested as thrombocytopenia and thrombopathy, which are the altered synthesis of vitamin K-dependent coagulation factors, and dysfibrinogenemia. Cirrhosis is associated with both quantitative and qualitative platelet abnormalities. Approximately 40% of cirrhotic patients have a prolonged bleeding time in excess of 10 minutes, and a corresponding decrease in platelet count to less than 100,000/mL10). The severity of the thrombocytopenia increases with a higher Child-Turcotte-Pugh score, in addition to platelet counts of less than 50,000/mL, which are commonly observed11). This decrease in the number of circulating platelets is related in part to pooling of platelets in the spleen caused by portal hypertension and splenomegaly which is in turn due to immunologic destruction of platelets12). In addition, thrombocytopenia occurs in alcoholic patients, which causes a direct suppression of bone marrow by alcohol.
Cirrhosis of the liver is also associated with functional abnormalities including, circulating platelets which are not activated in a normal manner resulting in defective clot formation13, 14). The decrease in platelet aggregation, as measured by the ristocetin test, may be related to decreases in glycoprotein 1b levels in the platelet membrane15) or to defective signal transduction within the platelets16).
With the loss of functioning hepatocytes, clotting factor deficiencies develop and may become quite severe. Moreover, vitamin Kdependent factors including, II, VII, IX, and X, as well as proteins S and C are affected early17). After its synthesis, vitamin K serves as a cofactor for a hepatic carboxylase which modifies these factors to provide a site for calcium binding, which is an essential step in their function. As factor VII levels decrease, there is a progressive increase in the prothrombin time, reflecting decreased function of the extrinsic coagulation cascade; however, factor VIII levels are normal.
Dysfibrinogenemia represents an activation of fibrinolysis and is detected by increased blood levels of fibrin degradation products, d-dimer, and tissue plasminogen activator in the presence of cirrhosis. Fibrinogen levels may be normal, but the thrombin and reptilase time are generally prolonged. Enhanced fibrinolysis increases the risk of gastrointestinal hemorrhaging18), and presumably other types of bleeding as well. In more severe cases, dysfibrinogenemia may progress to disseminated intravascular coagulation. Past studies indicate that the plasma concentration of antithrombin III appears to be important in this regard19). Thrombin formation is increased in cirrhosis, but is controlled if sufficient antithrombin III is present. If the levels of antithrombin III are low (<0.3 U/mL), thrombin is not inactivated, which in turn may lead to sustained interactions with fibrin and an increased risk of disseminated intravascular coagulation19).
Our case study belonged to Child-Pugh-Turcott score C and had a prolonged prothrombin time, partial thromboplastin time, and low platelet count; however, no traumas were added in the hospital. The patient died despite embolizations of the lateral thoracic artery and best supportive care. We suggest that cirrhosis of the liver itself, may develop anatomical or microstructural defects of some endothelial cell covered vessels with decreasing liver function, and that anatomically distorted vessels may bleed without trauma. Another possibility is the several clotting abnormalities of cirrhosis in combination with a possible underlying defect of the artery; whether it is congenital or acquired, may cause bleeding in response to the deterioration of liver function.
If patients with cirrhosis of the liver experience bleeding from any artery, it is necessary to implement more intensive and meticulous management. Early recognition of symptoms may help the treating physician reduce the likelihood of a catastrophic outcome. This is, to our knowledge, the first case of spontaneous rupture of the lateral thoracic artery in patients with liver cirrhosis.



 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print





