


INTRODUCTION
Myelodysplastic syndrome (MDS) is a heterogeneous group of clonal hematologic diseases characterized by ineffective hematopoiesis in the bone marrow (BM) and peripheral cytopenia, with the potential to progress to acute myeloid leukemia or fatal BM failure [1]. A key pathophysiologic process in MDS is accumulation of genetic damage. Mutation of genes that regulate critical cellular functions can drive malignant transformation [2]. The resulting unopposed activation and proliferation of immune cells might ultimately disrupt normal immune responses in MDS patients and contribute to development of autoimmune disease (AID). Indeed, approximately 20% of MDS patients develop AIDs [3], suggesting that MDS-associated AIDs might result from similar genetic changes.
Previously, we demonstrated that certain AIDs in MDS patients are associated with distinctive karyotypic abnormalities and outcomes [4]. While a karyotypic abnormality indicates a radical change in gene dose (with duplication or deletion of large gene segments), genetic modification without karyotypic abnormality can still drive development of MDS. Mutation of these ŌĆ£malignancy-associated genesŌĆØ contributes to AIDs. One critical mutation may involve proteins that regulate epigenetic modifications, which in turn impact cellular function. For example, mutation of ten-eleven translocation 2 (TET-2) is associated with both MDS and exacerbated inflammatory responses [5].
Here, we investigated whether particular gene mutations frequently associated with myeloid malignancy increase the risk of an AID developing in patients with MDS.
METHODS
Patients
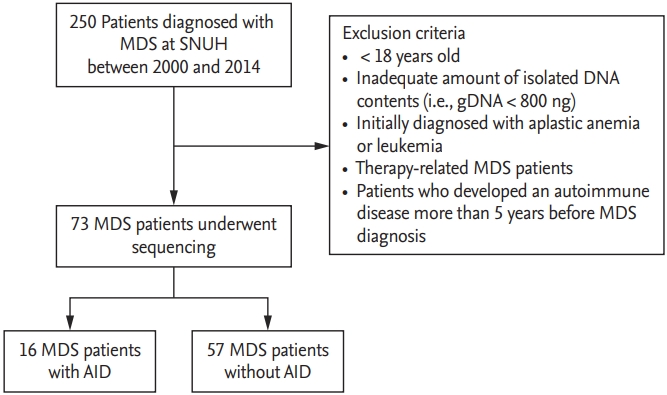
A total of 250 patients initially diagnosed with MDS at Seoul National University Hospital from January 2000 to December 2014 were included. Exclusion criteria were as follows: age at the time of diagnosis < 18 years, initial diagnosis of aplastic anemia or leukemia, and therapy-related MDS. Patients who developed an AID more than 5 years before MDS diagnosis were excluded. Patients from whom adequate amounts of DNA (i.e., Ōēź 800 ng) could not be extracted were also excluded. A final cohort of 73 treatment-naive patients was included in the analysis (Fig. 1). Laboratory and clinical data were obtained at time of BM sampling for gene sequencing. MDS subtypes were defined according to the World Health Organization classification [6]. The study was approved by the Institutional Review Board of Seoul National University Hospital and conducted in accordance with the Declaration of Helsinki (IRB protocol number: 1604-082-754). Written informed consent was obtained from all patients.
Definition of AID
Medical records were reviewed to evaluate the presence of an AID. Diagnosis of an AID was based on the 2010 American College of Rheumatology/Annual European Congress of Rheumatology criteria for rheumatoid arthritis (RA) [7], the 1984 modified New York criteria for ankylosing spondylitis [8], and the Bohan and Peter [9] criteria for idiopathic inflammatory myopathies. Autoimmune thyroiditis was defined as abnormal thyroid function in the presence of anti-thyroid antibodies (i.e., anti-thyroid peroxidase antibodies and anti-thyroid stimulating hormone receptor antibodies). Immune thrombocytopenia (ITP) was defined as thrombocytopenia in the presence of antiplatelet antibodies after excluding other causes of thrombocytopenia. Hemolytic anemia (HA) was defined as anemia in the presence of a positive direct Coombs test and laboratory evidence of hemolysis (such as increased lactate dehydrogenase levels and decreased haptoglobin levels). Panniculitis, Sweet syndrome, psoriasis, and uveitis were diagnosed clinically by consulting dermatologists and ophthalmologists, respectively.
BM histology
Wright-stained BM smears and hematoxylin and eosin-stained sections of BM trephine biopsies were reviewed by hematopathologists.
Targeted sequencing of 88 MDS-associated genes
Eighty-eight genes associated with myeloid malignancy were selected for targeted sequencing (Supplementary Table 1) [10]. To construct a sequencing library, genomic DNA (gDNA) was extracted from BM cells using the QIAamp DNA Blood Mini Kit (Qiagen, Valencia, CA, USA). A total target length of 259 Kb regions, with paired-ends of 150 bp, was sequenced on an Illumina HiSeq 2500 platform (Illumina, San Diego, CA, USA) using the rapid-run sequencing mode. The mean time between MDS diagnosis and gene sequencing was 4.4 days.
Processing of sequencing data and variant calling
A schematic diagram illustrating the data processing pipeline and the algorithm used for variant calling of somatic mutations is shown in Supplementary Fig. 1. The raw data were mapped to the reference genome (hg19) using the Burrows-Wheeler Aligner (BWA, v0.62). Duplicate PCR reads were removed using Picard 1.98, and variants were called using ŌĆ£UnifiedGenotyperŌĆØ in GATK 2.7-2. Variants flagged as ŌĆ£LowQualŌĆØ and low depth (< 10) were excluded. Variants were annotated using ANNOVAR and candidate gene mutations were selected according to the criteria described in the Supplementary Table 1. All synonymous variants were discarded. Missense single nucleotide variants (SNVs) reported in public databases, including single nucleotide polymorphism database (dbSNP), ESP6500, and the 1,000 genomes project, with a frequency of > 0.5% were filtered as polymorphisms. In addition, an in-house database of Korean polymorphisms, derived from the sequence data of 273 normal Koreans, was used as a private database to filter polymorphisms. SNVs that were found in more than two persons in the Korean database (allele fraction Ōēź 0.2%) were filtered out. From the filtered SNVs with low depth, SNV variants known from previous MDS studies to be hotspot mutations were rescued [11,12] using the criterion of > 5 total reads with supporting reads. Mapping errors were confirmed by visual inspection using the IGV browser. Among missense SNVs not reported previously, SNV mutations predicted to be benign by SIFT and Polyphen software were removed.
Statistical analysis
Continuous variables were expressed as the mean ┬▒ standard deviation or median (interquartile range [IQR]), and categorical variables were expressed as a number (%). StudentŌĆÖs t test and the chi-square test were used to compare continuous or categorical variables between the two groups. A p values Ōēż 0.05 were considered statistically significant. All statistical analyses were performed using SPSS version 23.0 (IBM Co., Armonk, NY, USA).
RESULTS
Baseline characteristics of MDS patients with or without AID
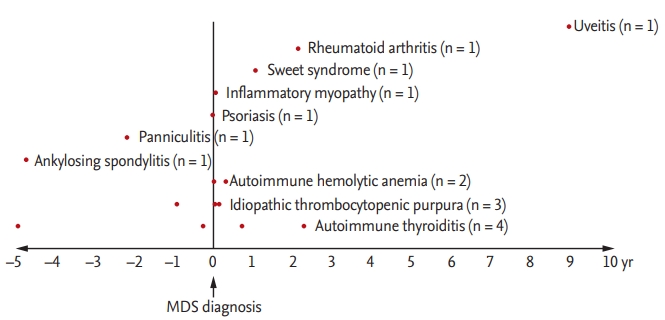
The median age of the 73 MDS patients was 70 years (IQR, 56 to 75), and 49 patients (67.1%) were male. The median follow-up duration after MDS diagnosis was 1.8 years (IQR, 0.9 to 3.3). Among the 73 patients, 16 (21.9%) were diagnosed with an AID: four (25%) with autoimmune thyroiditis, three (18.8%) with immune thrombocytopenia, two (12.5%) with HA, one (6.3%) with RA, one (6.3%) with panniculitis, and one (6.3%) with idiopathic inflammatory myopathy (Fig. 2). The baseline characteristics of MDS patients with or without an AID are shown in Table 1. The two groups did not differ in terms of sex, age, MDS subtype, and laboratory parameters.
The median time between diagnosis of MDS and AID was 0.03 years (IQR, ŌĆō0.8 to 1.0). Most AIDs were diagnosed shortly before or after MDS diagnosis. One patient developed uveitis 9 years after MDS diagnosis, and two patients (one RA and one autoimmune thyroiditis) developed AID 2 years after MDS diagnosis. The AIDs were treated with corticosteroid with or without immune suppressants. The case of autoimmune thyroiditis was treated with methimazole. All patients improved after treatment.
TET2 mutation is associated with increased risk of autoimmunity in MDS patients
The 88 selected genes sequenced herein are listed in Supplementary Table 1. Fifty-seven of 73 MDS patients (78.1%) harbored a genetic mutation in at least one of the selected genes. The presence of a mutation (68.8% vs. 80.7%, p = 0.32) and the mean number of mutations (1.8 ┬▒ 1.6 vs. 2.2 ┬▒ 1.8, p = 0.34) did not differ between the AID and non-AID groups. Strikingly, the rate of TET2 mutation was significantly higher in MDS patients with an AID than in those without (31.3% [n = 5/16] vs. 5.3% [n = 3/57], respectively; p = 0.001). The patients with TET2 mutation were older than those without (75 [70.8ŌĆō84] vs. 68 [55.5ŌĆō73], p = 0.02). However, the median age did not differ between the AID and non-AID patients (66.5 years [56.3ŌĆō72.5] vs. 70 years [56ŌĆō75], p = 0.66) (Table 1).
Logistic regression analysis revealed that TET2 mutation was associated significantly with an increased risk of AID (odds ratio [OR], 8.18; 95% confidence interval [CI], 1.70 to 39.38; p = 0.009) (Table 2). All observed TET2 mutations were variants of strong clinical significance, including stop-gain and frameshift deletion and insertion mutations (Table 3).
DISCUSSION
To the best of our knowledge, this study is the first to examine the potential effects of mutations in 88 selected myeloid malignancy-associated genes on development of AID in patients with MDS. Mutation of TET2, an epigenetic regulator, was the single most significant factor that increased the AID risk in MDS patients.
The prevalence of AID in the general population is about 3% to 5%, whereas that in MDS patients ranges from 10% to 30% [13-15]. Consistent with this, 16 (21.9%) of the patients with MDS in the present study developed an AID. This tight association between MDS and AID indicates that they may share common pathophysiologic mechanisms, including genetic and/or epigenetic modifications. We and others reported that particular karyotypic abnormalities are associated with specific AIDs in patients with MDS. For example, trisomy 8 is associated with Beh├¦et disease in MDS patients [4,16], whereas del (5q) is common in MDS patients with RA [17]. However, karyotypic abnormalities represent a substantial quantitative alteration in whole genome material, and 40.3% of MDS patients without an abnormal karyotype still develop an AID [4]. Here, we focused on the association between AID and gene mutations that are critical for development of hematologic malignancy [18]. As expected, 57 of 73 MDS patients (78.1%) harbored mutations in Ōēź 1 of 88 selected genes associated with hematologic malignancy. Strikingly, only the TET2 mutation was enriched significantly in patients with an AID compared with those without. Subgroup analysis of 43 MDS patients without a karyotypic abnormality revealed that three (30%) of ten patients with an AID harbored a TET2 mutation compared with three of 33 patients (9%) without an AID (p = 0.04) (data not shown). Therefore, alterations at the gene level might be critical for autoimmunity in MDS.
TET2 belongs to the TET family and plays a role in converting 5-methyl-cytosine (5-mC) to 5-hydroxymethyl-cytosine (5-hmC) during DNA demethylation [19]. Accordingly, loss-of-function TET2 mutations lead to decreased TET2 enzymatic activity, which is associated with changes in methylation profiles and epigenetic dysregulation [19]. TET2 deletion is sufficient to initiate myeloid and lymphoid transformation [20]. TET2 controls cytokine production by T cells in AIDs [21]. In addition, TET2 is a crucial negative transcriptional inhibitor of proinflammatory responses, and TET2 depletion contributes to accelerated atherosclerosis via increased inflammasome responses [22]. Accordingly, aberrant methylation of specific gene promoters might be associated with development of hematopoietic malignancy and autoimmunity [20]. In line with this, all TET2 mutations identified herein were variants of strong clinical significance and associated with presumably impaired TET2 function. However, a large prospective study is needed to investigate how TET2 mutation contributes to autoimmunity in MDS.
Previous studies report a strong association between TET2 mutations and aging [23,24]. Since TET2 mutations accumulate with age, it might be tempting to speculate that particular AIDs, especially those occurring later in life (e.g., polymyalgia rheumatic and giant cell arteritis [GCA]), might be, at least in part, caused by accumulating TET2 mutations [25,26]. A preliminary study revealed that TET2 mutation was not observed in GCA patients without MDS, suggesting that TET2 mutation contributes to development of AID in MDS patients but not in the general population (unpublished data).
The study has several limitations. First, the number of enrolled patients was relatively small, which may affect robust statistical analysis (e.g., correction for multiple comparisons). Second, the temporal relationship between TET2 mutation and AIDs development in patients with MDS could not be investigated precisely since the exact time of TET2 mutation could not be determined. Third, AID cases diagnosed within 5 years before MDS diagnosis were included, since chronic smoldering cases of MDS might remain undiagnosed for many years until emergence of obvious clinical symptoms. However, it is possible that those cases might not be related to MDS. Fourth, ITP in three patients was defined as isolated thrombocytopenia in the presence of antiplatelet antibodies after excluding other causes of thrombocytopenia. However, it is difficult to differentiate ITP from dysplastic megakaryopoiesis with a high degree of certainty. In addition, one patient with autoimmune thyroiditis after allogenic peripheral blood stem cell transplantation was included as an MDS-related AID. Since hematopoietic cell transplantation might clear MDS clones that might induce GraveŌĆÖs disease, GravesŌĆÖ disease occurring after hematopoietic cell transplantation might not be related to MDS; rather, it could be a manifestation of graft versus host disease. Finally, it would be of particular interest to investigate whether disappearance of clones with TET2 mutations after treatment of MDS (e.g., with BM transplantation) is associated with improvement of autoimmune phenomena. Further studies are warranted to validate the current finding in independent cohorts.
In conclusion, this study is the first to show that mutation of TET2 may be associated with a significant increase in the risk of AID in patients with MDS. Large prospective and validation studies are needed to identify more precise relationships and underlying mechanisms.
KEY MESSAGE
1. Autoimmunity is common in myelodysplastic syndrome (MDS).
2. Mutation of ten-eleven translocation 2 (TET-2), an epigenetic regulator, is associated significantly with increased risk of autoimmunity in MDS.
3. Particular genetic alterations might contribute to both autoimmunity and malignancy in MDS.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Supplement 1
Supplement 1 Print
Print





