


INTRODUCTION
Blastic plasmacytoid dendritic cell neoplasm (BPDCN), previously referred to as blastic natural killer cell lymphoma or CD4+/CD56+ hematodermic neoplasm, is a rare hematologic malignancy that is derived from plasmacytoid dendritic cells (PDCs) [1-4]. BPDCN, which was categorized as an “acute myeloid leukemia and related precursor neoplasm” by the World Health Organization (WHO) in 2008, is typically characterized by CD4+ and CD 56+ co-expression without common lymphoid or myeloid lineage markers, suggesting derivation from PDC precursor [1,3,5]. Since the first case was described, approximately 200 cases have been reported due to its extremely low incidence [6-8]. This disease has a tendency to involve the skin, lymph nodes, peripheral blood, and bone marrow [7,9], and the cutaneous involvement is a predominant clinical feature of BPDCN ranging in appearance from small bruise-like areas to patches, nodules, and ulcerated masses [3,9,10]. In spite of its indolent nature confined to the skin without any constitutional symptoms, radiotherapy was found to be ineffective because most patients eventually relapse [11]. Although a substantial number of patients respond to initial chemotherapy like lymphoma patients, the outcome of systemic chemotherapy also has been disappointing because of its relatively short duration of response [9,10,12]. As a result, the optimal treatment strategy for BPDCN has not been established because the outcome of various chemotherapy regimens is not satisfactory. As a current clinical practice, intensified chemotherapy regimens have been used as a primary treatment for BPDCN in conjunction with the consolidation treatment such as autologous or allogeneic stem cell transplantation (SCT). However, there are limited data on the outcomes of these treatment approaches, especially Asian patients with BPDCN [13-15]. Herein, we reported our single-center experience with BPDCN patients receiving various chemotherapy regimens.
METHODS
Patients
We queried the terms ‘blastic plasmacytoid dendritic cell neoplasm’ and ‘blastic natural killer cell lymphoma’ in biopsy reports in the Samsung Medical Center electronic medical records database from January 1, 2000 to June 30, 2014. Through this process, adult patients (age ≥ 18 years) whose medical records contained these terms were selected. After we manually reviewed the medical records of these patients, we selected 10 patients who were consecutively diagnosed with BPDCN. Those cases were reviewed by an expert pathologist for lymphoma according to the diagnostic criteria of the WHO 2008 classification [5]. Morphological analysis of skin and breast biopsies was conducted. All immunostains were performed on formalin-fixed, paraffin-embedded specimens. Immunohistochemical (IHC) criteria for diagnosis of BPDCN were expression of CD4 and CD56 by blastic tumor cells, coupled with at least on plamacytoid dendritic cell-specific antigen among CD123 or terminal deoxynucleotidyl transferase (TdT), in the absence of lineage-specific markers for B cells (CD20), T cells (CD3), monocytes (lysozyme), and myeloid cells (myeloperoxidase [MPO]). Clinical data obtained included sex, age, initial presentation, biopsy site, initial manifestation, laboratory findings at diagnosis, treatment, and outcomes. This retrospective study was approved by the Institutional Review Board of Samsung Medical Center (IRB No: 2015-12-021) for the release of case information that was rendered anonymous. Thus, obtaining an informed consent was waivered.
Treatment review and survival analysis
Since 2000, various chemotherapy regimens have been used for BPDCN at our institution according to physician’s preference. The most commonly used regimen was the hyperCVAD regimen (course A: cyclophosphamide, vincristine, doxorubicin, dexamethasone; course B: methotrexate, cytarabine) chemotherapy, and L-asparaginase-containing chemotherapy regimens including VPDL (vincristine, methylprednisolone, daunorubicin, L-asparaginase), and VIDL (etoposide, ifosfamide, dexamethasone, L-asparaginase). Thus, we compared patients according to the use of anthracycline-based intensified regimen and L-asparaginase-containing intensified regimen. Because all patients showed relapse or progression after their initial treatments, salvage chemotherapy regimens were also used including ESHAOX (etoposide, methylprednisolone, oxaliplatin, cytarabine), GDP (gemcitabine, dexamethasone, cisplatin), and SMILE (methotrexate, etoposide, ifosfamide, dexamethasone, L-asparaginase). We also analyzed the response to these salvage treatments based on the relevant response criteria. Thus, for patients diagnosed with BPDCN after 2007, 18F-fluorodeoxyglucose (FDG) positron emission tomography/computed tomography (PET/CT) was used according to the revised response criteria for malignant lymphoma whereas the response of patients was evaluated by the International Working Group criteria before 2007. Autologous or allogeneic SCT were also performed as a consolidation treatment for responders to chemotherapy after complete response (CR) to initial therapy or salvage treatment if patients were eligible for SCT. The last survival status update was done at the time of analysis in September 2015. Survival curves were generated by the Kaplan-Meier method and compared using the log-rank test. Overall survival (OS) was calculated from the date of diagnosis to the date of death resulting from any cause. The time to progression was the date of diagnosis to the date of documented disease progression or relapse.
RESULTS
Patient characteristics
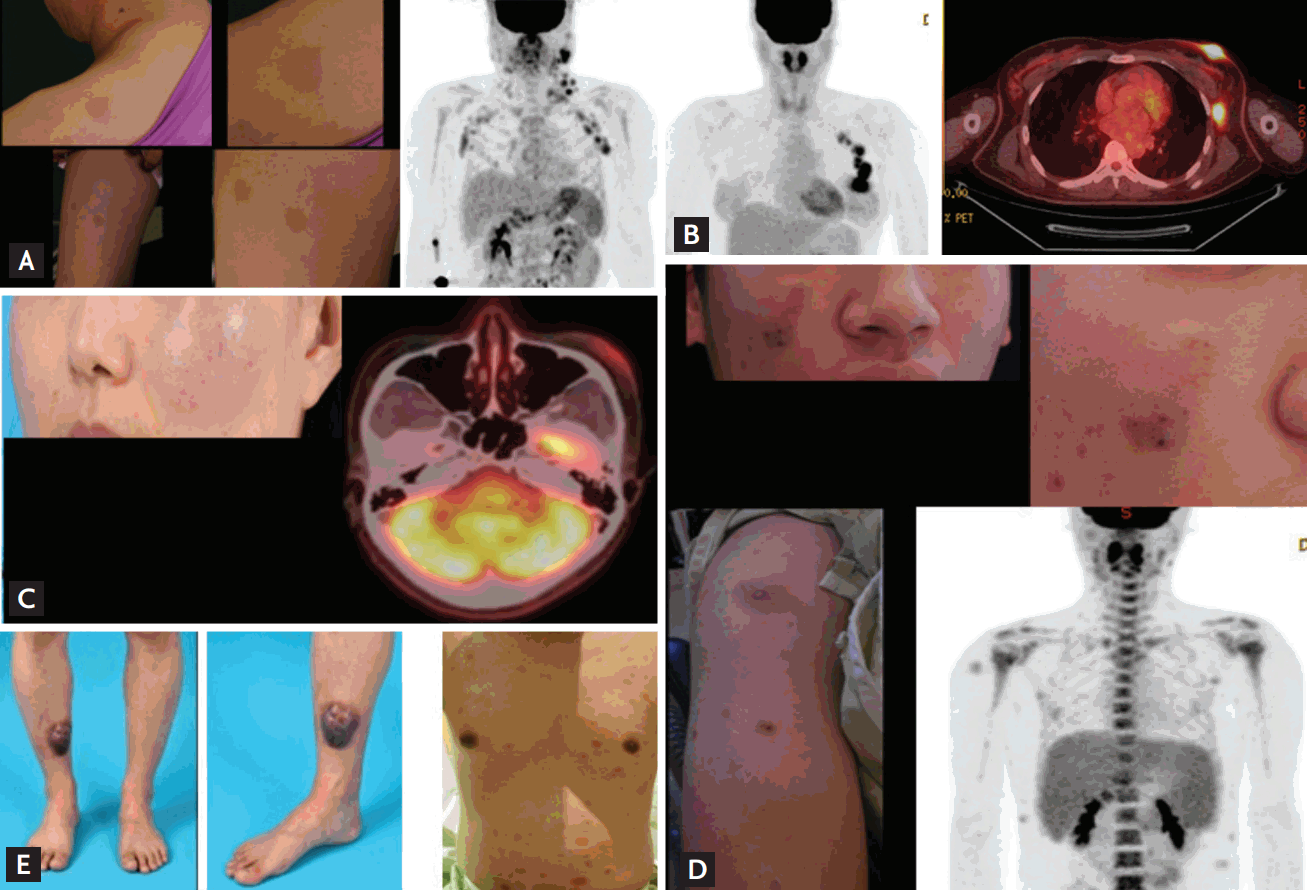
The study included seven male and three female patients with a median age at diagnosis of 41 years (range, 18 to 79). All patients had single or multiple skin lesions at presentation, and half of patients had multiple lymph node enlargements (Table 1). Cutaneous lesions were morphologically variable and involved the face, trunk, and extremities (Fig. 1). Six patients had a solitary cutaneous lesion, while the other patients had multiple skin lesions (Table 1). The median time between the onset of lesions and pathologic diagnosis was 2.6 months (range, 0.7 to 6.6). Bone marrow involvement was found in three patients at diagnosis, and they all showed abnormal findings in peripheral blood (Table 1). Two patients had hepatosplenomegaly, and both presented with thrombocytopenia. Only one patient (#4) experienced B symptoms including weight loss and chills at diagnosis. Half of patients showed elevated levels of serum LDH; however, other laboratory values including creatinine and albumin were within normal range. All but one patient were diagnosed with BPDCN without any relevant disease history. One patient (#9) previously had myelodysplastic syndrome (refractory anemia with excess blasts-2), and received decitabine treatment followed by allogenic hematopoietic SCT with fludarabine and busulfan conditioning. Twenty-two months later, he presented with cheek swelling and was diagnosed with BPDCN.
Pathology
For pathologic diagnosis, seven patients underwent skin biopsy, one patient underwent breast mass and axillary lymph node biopsies, one lymph node biopsy, and one periorbital and maxillary mass biopsies. On immunohistochemistry, patients showed CD4, CD56, and CD123 positivity, and negativity of CD3, CD20, and MPO in tumor cells (Table 2). Conventional cytogenetic studies with bone marrow aspirates at diagnosis indicated that three patients had abnormal cytogenetics (Table 2). The findings of 18F-FDG PET/CT were available in seven patients. Of these patients, the FDG uptake of lesions were variable, and this uptake was minimal in patients with only cutaneous manifestations. The percentage of Ki-67 was relatively low in most cases, similar to indolent lymphomas.
First-line treatment and outcome
Four patients received hyperCVAD chemotherapy as a first-line treatment, and three patients showed a CR, whereas one patient showed a partial response (PR). Among them, the patient with a PR (#2) received adjuvant radiotherapy for breast mass and axillary lymph nodes. However, she developed local relapse 6 months later. Another patient (#7) with CR underwent upfront allogenic SCT from a fully matched sibling donor after treatment with 10 Gy total body irradiation conditioning and cyclophosphamide 60 mg/kg body weight. He maintained a CR for 14 months, but ultimately relapsed with multiple skin lesions. In particular, leukemic transformation was found in one patient (#1) at the time of relapse 19 months after first-line treatment (Table 3). One patient initially treated with CHOP (doxorubicin, cyclophosphamide, vincristine, prednisone) also showed disease progression after she achieved PR (#10). Three patients received L-asparaginase-containing chemotherapy regimens including VPDL and VIDL. Although they all responded to the treatment, they all relapsed (#3, #5, and #6) (Table 3). One patient (#9) received palliative radiation therapy to the maxilla, but his periorbital mass progressed. The other patient (#4) did not receive any treatment with curative intent because of advanced age and comorbidity.
Salvage treatment and outcome
After relapse or disease progression occurred, second-line treatment with combination chemotherapy, local radiotherapy, or autologous SCT were applied. The regimens used included ESHAOX, GDP, VPDL, and SMILE. Most patients responded poorly to second-line therapies; however, one patient (#8) was salvaged with four cycles of ESHAOX, and underwent autologous SCT. Although he maintained the 2nd CR for 7 months, he relapsed and died due to disease progression (Table 3). Another patient (#1) achieved a PR after completion of the 1st cycle of VPDL; however, he immediately progressed to leptomeningeal involvement and subsequently died. Interestingly, patient #2 who presented with a breast mass and multiple involved lymph nodes received SMILE, DHAP (cisplatin, cytarabine, dexamethasone), and GDP as salvage chemotherapy, but she did not respond to treatment. Finally she underwent intensified chemotherapy with MEC (mitoxantrone, etoposide, cytarabine), and remains in CR without any signs of relapse.
Survival outcome
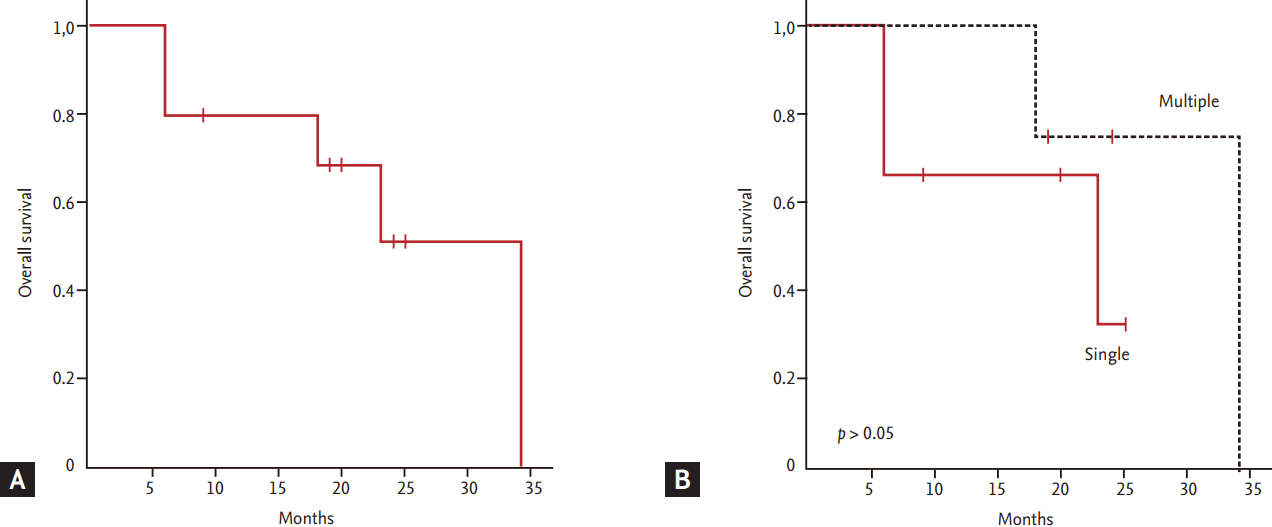
The median OS of patients was 34 months, and five patients were alive at the time of analysis (Fig. 2A). All patients experienced at least one relapse event or progression regardless of whether they had a single or multiple skin lesions at diagnosis, with a median progression-free survival of 11.2 months (range, 6.2 to 19.4). The pattern of relapse was also variable; thus, some patients showed repeated local relapse (e.g., patient #2), while other patients showed distant relapse (e.g., patient #1 and #5). Thus, the initial presentation of skin lesions was not significantly associated with OS (Fig. 2B). Although one patient who underwent SCT did not show evidence of relapse, she died due to transplantation-related complications (pneumonia and chronic graft-versus-host disease).
DISCUSSION
This rare but fatal disease entity was first found to originate from the precursor of PDCs in 1999, and the final diagnosis criteria for BPDCN were determined in the 2008 version of the WHO classification of tumors of hematopoietic and lymphoid tissue [16,17]. A previous study analyzing the cutaneous presentation of 90 patients with BPDCN described 66 patients (73%) who presented with nodular lesions, 11 patients (12%) with ‘bruise-like’ patches, and 13 (14%) with disseminated lesions (patches and nodules) [10]. Consistent with these findings, all patients in our study had cutaneous lesions including two patients with nodules, four with macules, and two with masses. Pathologically, BPDCN shows abundant cytoplasm with a low or medium nucleus to cytoplasmic ratio and displays basophilia without granulation; diagnosis is based on the immunophenotypic profile [9]. Given its rarity and only recent recognition as a distinct clinicopathologic entity, there is no consensus on the immunohistochemistry markers for diagnosis of BPDCN [9,10]. However, a previous study of 91 patients using a panel of 12 IHC markers identified five markers (CD4, CD56, CD123, CD103, and TCL1) that could be important for diagnosis [10]. Thus, a typical case of BPDCN could co-express CD4, CD56, and CD123 without expressing common lymphoid or myeloid lineage markers [8,18]. A karyotyping study demonstrated that cytogenetic changes were complex, mainly involving chromosomes 5, 9, 12, 13, and 15 [19]. In our study, all patients expressed CD4, CD56, and CD123. Among these, eight patients did not express common lymphoid or myeloid lineage markers such as CD3, MPO, and lysozyme. Only lysozyme stained positively in 5% of tumor cells in one patient. Cytogenetic abnormalities were observed in three patients, while seven patients displayed a normal karyotype.
Because there is no consensus on the optimal therapy for BPDCN, therapeutic approaches vary widely from radiation therapy in localized stages to myeloablative therapy. Reimer and colleagues [20] reported that a CHOP-like regimen was used most frequently, and the overall response rate in 38 patients was about 71% (CR, 55%; PR, 16%). However, accumulated data showed that radiation therapy or CHOP-like regimens failed to sustain CR status [7,11]. Dalle et al. [11] reported that CHOP-like regimens frequently induce disease remission; however, the duration of response was only around 5 months. In general, ALL/lymphoma-type regimens including hyperCVAD and VPDL were reported to show better survival outcomes than AML-type regimens [12,21]. Thus, in a previous study of 41 patients, the CR rates of ALL/lymphoma and AML-type regimens were 67% and 27%, respectively (p = 0.02). In our study, seven patients who received hyperCVAD or VPDL showed an objective response (five patients, CR). Responses were relatively long-lived, with a median length of 11.2 months (range, 6.2 to 19.4), compared with those previously reported for CHOP-like regimens [20]. Nevertheless, all patients showed relapse or disease progression (Table 3). Thus, regimens such as hyperCVAD and VPDL did not appear to be sufficient to ensure durable long-term remission. Allogenic SCT with myeloablative conditioning (MAC) has been shown to induce durable disease control leading to cure [11,20]. A study analyzing 34 patients who underwent allogenic SCT reported a 3-year disease-free survival and OS of 33% and 41%, respectively. They also reported that long-term disease control was only observed after MAC, suggesting that MAC allogeneic SCT is an effective treatment. However, it is not clear to what extent intensive induction chemotherapy contributed to the success of the above study. Because BPDCN occurs in patients of advanced age and/or with comorbidities, MAC conditioning might not be suitable for elderly patients. A few case series described the benefit of autologous SCT or reduced intensity conditioning allogeneic SCT [21,22]. A recent study comparing 11 patients who underwent autologous SCT and 14 patients who underwent allogeneic SCT in CR1 showed that the OS rates at 4 years for patients who underwent autologous SCT and allogeneic SCT were 82% and 53% (p = 0.11) with a median follow-up of 53.5 months [15]. Thus, high-dose chemotherapy itself might provide a survival benefit for patients with BPDCN. In our study, one patient (#2) with repeated relapses was able to maintain a CR after intensified chemotherapy, MEC (Table 3). However, the role of autologous SCT should be elucidated compared to allogeneic SCT.
The prognosis of patients with BPDCN is very poor with a median OS of about 12 to 14 months [9,10,12,20]. Several prognostic factors, including presentation with only localized skin lesions, Ki-67 proliferative index, TdT, and biallelic loss of locus 9p21, have been proposed in terms of survival [10,20,23]. In our study, two patients with only cutaneous involvement maintained longer remission compared to patients with involvement of multiple lymph nodes; however, patients presenting with only localized cutaneous lesions did not exhibit superior OS. Whether patients with only cutaneous lesions exhibit a better response to chemotherapy and prolonged survival remains unclear. In our cases, Ki-67 and expression of TdT were not related to response to first-line chemotherapy or OS. Herein, we summarized the clinical features and outcomes of 10 patients with BPDCN. Isolated cutaneous lesions are frequently the first symptom, but they eventually disseminate. All patients treated with hyperCVAD or VPDL showed an objective response, but the above regimens do not appear sufficient for inducing long-term remission. Although the small number of patients in our study precludes drawing definitive conclusions, the development of more effective induction as well as consolidation treatment strategy should be warranted to improve this rare disease entity.
KEY MESSAGE
1. BPDCN (blastic plasmacytoid dendritic cell neoplasm) is rare and fetal disease, and there is no consensus on the optimal therapy.
2. Intensified chemotherapy with or without stem cell transplantation showed an objective response, but the duration of response was relatively short.
3. More effective induction as well as consolidation treatment strategy should be warranted.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print





