


INTRODUCTION
The cardiovascular disease-related mortality with diabetes mellitus is ~65%. Therefore, diabetes mellitus is regarded as a risk equivalent to coronary heart disease [1]. Diabetic heart disease is a growing and important public health risk [2]. It affects the heart in three ways: cardiac autonomic neuropathy (CAN), coronary artery disease (CAD) due to accelerated atherosclerosis, and diabetic cardiomyopathy (DCM) [1]. DCM is characterized by lipid accumulation in cardiomyocytes, fetal gene reactivation, and left ventricular (LV) hypertrophy, which together result in contractile dysfunction [2].
In 1881, Leyden first reported that DCM is a typical complication of diabetes mellitus. In 1888, Mayer asserted that diabetes mellitus is a metabolic disorder that can induce heart disease. Finally, the term “diabetic cardiomyopathy” was proposed by Rubler in 1972 after postmortem studies in diabetic patients with heart failure in whom coronary disease and other structural heart diseases, hypertension, and alcohol had been ruled out as possible causes [3]. A milestone study in 2002 by Finck and colleagues [4] cast light on the transcriptional mechanisms of DCM. These researchers suggested that the transcription factor, peroxisome proliferator-activated receptor (PPAR)-α, along with its transcriptional targets, is upregulated in the hearts of mouse models of diabetes mellitus [2,4]. Currently, DCM is defined as myocardial dysfunction (MD) in patients with diabetes mellitus in the absence of hypertension and structural heart diseases such as valvular heart disease or CAD [5].
Diabetes mellitus is a well-known risk factor for the development of heart failure. The Framingham Heart Study demonstrated that the frequency of heart failure is five times greater in diabetic women and two times greater in diabetic men compared with age-matched control subjects [6]. Heart failure leads to a poor quality of life in affected individuals and complicates the treatment of diabetes mellitus by altering the pharmacokinetics of anti-diabetic medications. Thus, both the prompt diagnosis and early management of these patients are of utmost importance. However, DCM is poorly understood by most physicians, even cardiologists and diabetologists. Therefore, in this review we focus on the pathophysiological mechanism behind DCM and management strategies, including emerging therapeutics and diagnostic evaluation.
EPIDEMIOLOGY OF DIABETIC CARDIOMYOPATHY
The prevalence of heart failure among diabetic patients was as high as 19% to 26% in various clinical trials [7,8]. These two disease entities tend to coexist, and the impact of each condition on the other has bidirectional influences in terms of causation and outcome [9-11]. The Framingham Heart Study reported that 19% of patients with heart failure have type 2 diabetes mellitus (T2DM) and that the risk of heart failure increases 2- to 8-fold in the presence of T2DM [10,12,13]. Furthermore, an increase of 1% in hemoglobin A1c (HbA1c) levels is related to an 8% increase in the risk of heart failure, independently of age, body mass index, blood pressure, and the presence of CAD. This suggests that the risk of heart failure is controlled by factors unique to T2DM, such as hyperglycemia and insulin resistance [10,12,13]. Conversely, a 1% reduction in HbA1c levels is related to a 16% reduced risk of developing heart failure and poor outcomes [10]. This bidirectional interaction has provided evidence to support the existence of DCM as a distinct clinical condition, and suggests that the presence of diabetes mellitus might independently increase the risk of heart failure [9].
The prevalence of DCM is not yet clear because of a lack of large study outcomes from different populations with diabetes mellitus. The prevalence of diastolic dysfunction in patients with T2DM was up to 30% in some studies [14]. However, there are other studies that reported a prevalence rate as high as 40% to 60% [15]. A recent major prospective study examining the prevalence of heart failure and MD in patients with chronic (≥ 10 years) type 1 diabetes mellitus (T1DM) showed a prevalence of 3.7% and 14.5%, respectively, at the end of a 7-year follow-up [16]. The annual incidence of heart failure and MD were 0.02% and 0.1%, respectively. Diastolic heart failure accounted for 85% of the cases of heart failure [1,16].
PATHOPHYSIOLOGICAL MECHANISMS OF DIABETIC CARDIOMYOPATHY
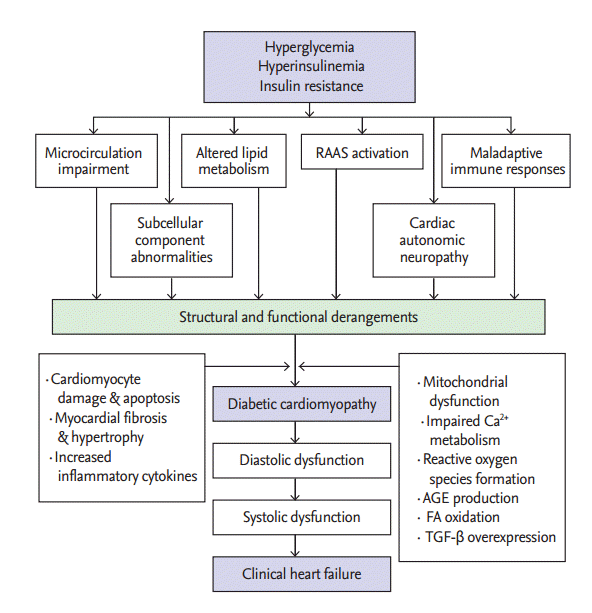
The pathophysiological mechanisms of DCM have not yet been sufficiently elucidated. The occurrence of DCM is multifactorial, and there are various proposed mechanisms including insulin resistance, microvascular impairment, subcellular component abnormalities, metabolic disturbances, cardiac autonomic dysfunction, alterations in the renin-angiotensin-aldosterone system (RAAS), and maladaptive immune responses (Fig. 1) [1,9,17].
A long-standing hypothesis is that hyperglycemia plays a pivotal role in the development of DCM, although multiple complex mechanisms and the interplay of many metabolic and molecular events within the myocardium and plasma contribute to its pathogenesis. The main metabolic abnormalities in diabetes mellitus are hyperglycemia, inflammation, and hyperlipidemia, all of which induce the production of the nitrogen species or reactive oxygen species (ROS) that cause most diabetic complications, including DCM and diabetic nephropathy [18].
Effects of hyperglycemia on the heart
Persistent hyperglycemia causes a number of molecular and metabolic changes in cardiomyocytes. Increased glucose metabolism due to hyperglycemia increases oxidative stress via the development of ROS from the mitochondria [19]. Oxidative stress caused by the overproduction of superoxide in the mitochondrial respiratory chain leads to reduced myocardial contractility and eventually induces myocardial fibrosis [20]. Oxidative stress and ROS can accelerate cardiomyocyte apoptosis and cellular DNA damage. Oxidative stress-induced DNA damage also activates DNA reparative enzymes such as poly ADP ribose polymerase (PARP) [21]. PARP redirects glucose metabolism from its usual glycolytic pathway to an alternative biochemical pathway that results in the development of various mediators and causes hyperglycemia-induced cellular injury. These injuries include increased hexosamine and polyol flux, protein kinase C activation, and advanced glycation end product (AGE) levels. The oxidative stress induced by persistent hyperglycemia increases the amount of AGEs in diabetic subjects [20]. AGEs can covalently crosslink various extra-and intracellular proteins that are thought to be important factors in diabetic complications. The crosslinking involving elastin and collagen results in impaired cardiac relaxation and increased myocardial stiffness. AGEs cause myocardial damage in both humans [22] and animals [23].
Contributions of insulin resistance and hyperinsulinemia
Insulin resistance and hyperinsulinemia are typical pathological abnormalities in T2DM and prediabetic conditions. Hyperinsulinemia leads to cardiomyocyte hypertrophy via various mechanisms. Cardiomyocyte hypertrophy in diabetes mellitus is modulated at the transcriptional level [24]. Various epigenetic and genetic alterations resulting from hyperinsulinemia activate multiple transcription factors that regulate extracellular and cellular protein expression. The activation of such transcription factors causes the deposition of extracellular matrix proteins and cardiomyocyte hypertrophy, resulting in focal myocardial fibrosis in diabetes mellitus [24].
Microcirculation impairment in the myocardium
The pathological characteristic of diabetes-related vascular complications is damage to the microcirculation throughout the body. Unique examples of microvascular complications inlcude diabetic nephropathy, neuropathy, and retinopathy [1].
Impaired coronary microvasculature is frequently observed in patients with T2DM, insulin resistance, and DCM [25,26]. This defect is caused by reduced levels of bioavailable nitric oxide [27]. In coronary vascular smooth muscle cells, nitric oxide activates both kinases and guanylyl cyclase, which is required for coronary relaxation [28]. Under conditions of reduced insulin sensitivity, both increased nitric oxide degradation and reduced nitric oxide production occur.
Reduced capillary length density and hyaline-related changes in the medial arteriolar layers are observed in the cardiac circulation of diabetes mellitus patients [17,29]. The reduced blood supply resulting from microcirculatory dysfunction affecting the vasa vasorum in diabetes mellitus further damages the medium and small arterioles of the diabetic heart. Perivascular fibrosis and interstitial changes, the formation of microaneurysms in small vessels, and thickening of the capillary basement membrane are other vascular disorders that cause cardiac microvascular ischemia in diabetes mellitus. Ischemia contributes to myocardial fibrosis, stiffness, and dysfunction in DCM.
Hyperinsulinemia and insulin resistance are associated with stiffness of both the small and large blood vessels [30]. Hyperinsulinemia promotes the differentiation of vascular smooth muscle cells to an osteoblast-like phenotype, which could play a role in the observed increased vascular stiffness [31]. Elevated insulin levels might also enhance vascular stiffness by increasing osteocalcin expression, alkaline phosphatase activity, and the formation of mineralized nodules in vascular smooth muscle cells via increased levels of receptor activator of nuclear factor κB [32]. Thus, impaired vascular smooth muscle cell and endothelial cell function are related to an increased risk of developing CAD in association with DCM.
Subcellular component abnormalities
Metabolic abnormalities involving endoplasmic reticulum (ER) stress, impaired calcium handling, and mitochondrial dysfunction are associated with the pathogenesis of DCM [11,33,34].
The overproduction of ROS exerts deleterious effects on the ER by impairing post-translational modifications and protein folding in the rough ER [25]. Stress to the ER induces an adaptive response that increases the proteasomeal degradation of incorrectly folded proteins [34].
Under conditions of excessive fat and carbohydrate intake and insulin resistance, nutrient overflow into cells causes the transfer of electrons to oxygen without adenosine triphosphate (ATP) production and also promotes a state of increased ROS, which potentially leads to oxidative damage within mitochondria [35]. Therefore, in the presence of mitochondria-produced ROS, DNA, proteins, and lipid membrane components are damaged and the accumulation of ROS-mediated fibrosis promotes diastolic dysfunction that can progress to heart failure.
ER and oxidative stress can impair calcium handling, which subsequently causes diastolic dysfunction and DCM. ROS, long-chain acylcarnitines, and abnormal membrane lipid content in the mitochondrial membrane, such as via cardiolipin, can alter calcium handling by affecting various transporter proteins; thus, inducing delayed diastolic relaxation and impaired intracellular calcium uptake [17,36]. The interactions between abnormal calcium handling, ROS, and ER stress enhance subcellular component dysfunction and ultimately lead to autophagy, necrosis, and apoptosis [26,37].
Effects of altered lipid metabolism on the myocardium
The heart can use both glucose and fatty acids (FAs) as energy substrates under physiological conditions. The uptake of FAs is mediated by FA translocase and the cluster of differentiation 36 (CD36), whereas glucose intake occurs via insulin-stimulated glucose transport, which is mediated by glucose transporter 4 (GLUT4) [11,38]. Nutrients promote myocardial insulin signaling and increase plasma insulin levels, which enhance the translocation of GLUT4 and CD36 to the myocyte sarcolemma to supply myocardial energy substrates [11]. In contrast, under conditions of T2DM and/or insulin resistance, GLUT4 is internalized and returns to its intracellular location and CD36 becomes preferentially localized to the sarcolemma [39].
The reciprocal positioning of CD36 and GLUT4 influences the genesis of cardiac metabolic abnormalities that are characterized by metabolic inflexibility [39]. Thus, the reduced glucose uptake caused by cardiac and systemic insulin resistance promotes a substrate shift toward increased free fatty acid (FFA) oxidation in diabetes mellitus, resulting in reduced cardiac efficiency [33,40]. The lipotoxicity characterized by the excessive accumulation of FAs in the myocardium reduces normal physiological autophagy and impairs insulin signaling, which leads to structural and morphological alterations and impaired myocardial performance [33]. These abnormalities facilitate myocardial oxygen consumption and can reduce the efficiency of muscle fiber function in response to electrical stimuli [17,36].
Overstrain of the cellular oxidation capacity in diabetes mellitus leads to ectopic lipid deposition in non-adipose tissues such as the heart, liver, and skeletal muscle. Recently, cardiac steatosis was proposed as an important cause of DCM [41,42]. The contribution of glucose oxidation to cardiac energetics is less than normal among patients with T2DM and obesity, and FA metabolism, instead, meets the myocardial energy needs [43]. Increased plasma FFA levels in patients with obesity and T2DM cause increased cardiac triglyceride accumulation and FA uptake. Excessive FA uptake and delivery by cardiomyocytes in this setting is likely to exceed the mitochondrial oxidative capacity and, thus, induces lipotoxic injury to the myocardium. Some of the excess FA enter nonoxidative pathways, leading to toxic FA intermediates such as ceramide. These toxic substances subsequently interfere with normal cellular signaling and give rise to apoptosis, cellular damage, mitochondrial dysfunction and, eventually, contractile dysfunction and myocardial fibrosis. Increased FA oxidation in the mitochondria is associated with the increased production of ROS, which oxidize cytoplasmic lipids into lipid peroxides. In turn, lipid peroxides and ROS lead to mitochondrial and cellular damage and the uncoupling of mitochondrial oxidative metabolism [44]. Consequently, the impaired myocardial generation of energy and reduced cardiac contractility are observed. Decreased energy production also induces impaired mitochondrial calcium handling, which leads to cardiac dysfunction [45]. Lipotoxicity-induced cellular apoptosis is generally referred to as lipoapoptosis. Different mechanisms may induce lipoapoptosis, such as palmitate toxicity, ER stress, diacylglycerol and ceramide formation, inflammation, and membrane destabilization [44]. The effects of lipoapoptosis on cardiac function include myocardial fibrosis and structural damage.
Cardiac autonomic neuropathy in diabetic cardiomyopathy
A previous study reported a relationship between the development of DCM and nervous system activation states [46]. Activation of the sympathetic nervous system increases β1-adrenergic signaling and expression, which facilitates interstitial fibrosis, cardiomyocyte hypertrophy, and impaired contractile function accompanied by increased cardiomyocyte apoptosis [17,36]. Activation of the parasympathetic nervous system is reduced, muscarinic receptor composition and density are altered, and acetylcholinesterase activity is decreased during heart failure [47]. Either indirect or direct stimulation of the vagus nerve might have direct beneficial effects on both cardiac remodeling and clinical outcomes [47].
CAN is a chronic complication of diabetes mellitus that leads to abnormalities in vascular hemodynamics and heart rhythm. The prevalence of varying degrees of CAN may be as high as 60% in individuals with a persistent history of diabetes mellitus [48]. CAN changes the contractile function of the myocardium and also influences blood flow in the coronary circulation. Patients with CAN exhibit increased peripheral vascular resistance and reduced vascular elasticity due to abnormal sympathetic tone [49]. A reduction in the myocardial perfusion reserve was also reported by other researchers [50]. This may partially account for the ventricular dysfunction associated with diabetic CAN.
Cardiac dysfunction is usual in diabetic patients with CAN [51]. Correlations between the prevalence of diastolic dysfunction and the severity of CAN have also been reported [51]. Alterations in the myocardial contractility responses associated with stress are observed in patients with diabetic CAN, and exercise-induced MD has been recorded even in patients with normal ventricular function at rest [52].
Activation of the renin-angiotensin-aldosterone system
Current evidence from human and animal experiments has supported a significant role for RAAS in diabetes-induced cardiac dysfunction [53,54]. Hyperglycemia activates the intracardiac RAAS, which exhibits different effects on cardiomyocytes. Intracellular angiotensin (AGT) II levels were 3.4-fold higher in the myocardial cells of diabetic patients compared with non-diabetics [55]. Cytoplasmic AGT II promotes cell growth in animal models. AGT II has a direct effect on cell signaling, leading to the proliferation of cardiac fibroblasts and cardiomyocyte hypertrophy [53]. Other factors, such as inflammation, aldosterone, and oxidative stress, may lessen the harmful effects of AGT II on the heart that cause myocardial damage in diabetes mellitus [54]. Moreover, the enhanced activation of mineralocorticoid receptor signaling and AGT II might facilitate insulin resistance by activating the mammalian target of rapamycin-S6 kinase 1 signal transduction pathway [56].
Maladaptive immune responses
DCM can be facilitated by alterations in the adaptive and innate immune systems [57,58]. The activation of macrophage polarization to classic (M1) or alternative (M2) phenotypes and proinflammatory T helper cells often occurs in states of insulin resistance or obesity [59]. Moreover, persistent overfeeding induces immune responses that contribute to low-grade inflammation within white adipose tissue [38]. Macrophage M2 polarization is an anti-inflammatory response and M1 polarization is a proinflammatory response in insulin-resistant and obesity states [59,60]. M1 macrophages secrete inflammatory cytokines, which reduce cardiac and systemic insulin signaling and facilitate the development of DCM [59]. In contrast, M2 macrophages secrete macrophage mannose receptor 1 and interleukin 10, which reduce the development of myocardial fibrosis and cardiomyocyte hypertrophy [59,61].
Another population of immune cells (T helper lymphocytes) was identified in patients with DCM [62]. A higher CD8+:CD4+ T-cell ratio is found in the visceral adipose tissue of mice fed a high-fat diet compared with lean mice [63,64]. Diet-induced insulin resistance also induces a dramatic increase in type 1 T helper-polarized cells, whereas the type 2 T helper-polarized fraction is reduced by ~50% [63]. These cells can be subtyped according to their cytokine expression profile following activation [64]. The increased secretion of chemokines, growth factors, and proinflammatory cytokines by T helper lymphocytes results in increased impaired diastolic relaxation and cardiac fibrosis [65]. However, regulatory T-cells usually attenuate the proinflammatory effects of T helper cells in the heart [66].
FUNCTIONAL AND STRUCTURAL ALTERATIONS
Significant alterations in myocardial function and structure occur as a result of DCM, leading to the pathological-clinical consequences of the disease. Pathological changes occur mostly in the myocardial interstitium in the early stages of the disease, and cardiac contractile dysfunction ensues as a consequence of these alterations [67]. Later, cardiac microcirculatory dysfunction, interstitial and perivascular fibrosis, and ventricular myocardial hypertrophy are observed.
Impaired diastolic function is the earliest abnormality in DCM, and systolic dysfunction develops only in the later stages of the disease [67,68]. DCM-induced ventricular fibrosis and hypertrophy are the major causes of diastolic dysfunction. When systolic dysfunction occurs, cardiac output declines progressively with the severity of the disease.
Interaction with coexistent coronary artery disease and hypertension
DCM is diagnosed only when CAD and hypertension have been ruled out. However, when these diseases overlap with existing DCM, the rapid progression to advanced heart failure may ensue. It is difficult to verify the role of these diseases in the progression and development of DCM from a clinical perspective [1].
Coexistent hypertension was observed in nearly 30% of patients with T1DM and in 50% to 80% of patients with T2DM in the United States [69]. Cardiac dysfunction was reportedly worsened by hypertension in animal models of DCM [70]. The presence of hypertension was independently associated with diastolic dysfunction in diabetic patients [71].
Cardiac remodeling in diabetic cardiomyopathy
DCM originates from the functional, structural, and regulatory remodeling of the heart caused by diabetes mellitus. Different stages of remodeling have been proposed, such as the early, advanced, and late stages [72].
In the early stage of DCM, metabolic disturbances such as hyperglycemia and insulin resistance are not accompanied by substantial alterations in myocardial systolic function and structure [36,37]. However, impaired myocardial relaxation can be detected by magnetic resonance imaging (MRI) and echocardiography. DCM is initially characterized by increased impairments in relaxation and cardiac stiffness with elevated atrial filling and reduced early diastolic filling [36]. Impaired insulin signaling also leads to a decrease in myocardial bloodflow reserve, which can be identified using various imaging techniques [73].
In the advanced stage of DCM, many alterations at the cellular level increase cardiac fibrosis, which induces substantial changes in systolic and diastolic function [17].
DIAGNOSIS OF DIABETIC CARDIOMYOPATHY
A number of DCM cases are subclinical, and patients may not have any overt signs or symptoms of the disease. In the early stages, there are only substructural cardiomyocyte changes, and detection is possible only using very sensitive methods such as strain rate, strain, and myocardial tissue velocity [17]. Subsequently, myocardial fibrosis and hypertrophy develop, which may be associated with structural changes such as increased myocardial mass and LV hypertrophy. Conventional diagnostic methods such as echocardiography may detect systolic and/or diastolic dysfunction at this stage. Significant fibrosis and microcirculatory changes occur in the myocardium in the advanced stages of DCM; this stage is usually associated with overt heart failure, ischemic heart disease, and hypertension [17]. Recently, several biomarkers have emerged for the detection of metabolic changes (Table 1).
Echocardiography
Echocardiography is a relatively inexpensive diagnostic method for assessing functional and structural cardiac abnormalities. Transmitral Doppler is the most commonn technique for evaluating LV diastolic function [75]. Tissue Doppler imaging (TDI) measures myocardial tissue velocities during the cardiac cycle and can be used to quantitatively estimate regional and global diastolic and systolic myocardial functions [76]. TDI is a more sensitive and specific diagnostic tool for detecting DCM compared with transmitral Doppler [77]. Fortunately, newer echocardiographic imaging techniques are currently evolving with better sensitivities and specificities.
Magnetic resonance imaging
Cardiac MRI recently emerged as a well-accepted imaging tool for the diagnosis of various functional and structural myocardial disorders [78]. Cardiac MRI is also useful for identifying myocardial steatosis and diastolic dysfunction [42]. Positron emission tomography (PET) and cardiac MRI using different radionuclides can help assess myocardial metabolic abnormalities; these newer imaging techniques may be helpful for diagnosing DCM.
Coronary angiography and cardiac catheterization
Cardiac catheterization is the best tool for evaluating the hemodynamic events within the heart chambers, since diastolic dysfunction documented invasively through catheterization continues to be the most definitive evidence of diastolic heart failure [79]. A mean pulmonary capillary wedge pressure > 12 mmHg or an LV end-diastolic pressure > 16 mmHg, determined invasively by catheterization, are the best diagnostic features of diastolic dysfunction [79]. However, the catheter-based diagnosis of DCM is rarely used because of the availability of highly sensitive and specific noninvasive techniques. Coronary angiography is helpful for detecting CAD that may coexist with or complicate DCM. Microvascular CAD is also detected by coronary angiography.
Serologic markers
Alterations in the levels of various serum/plasma cardiac biomarkers may reveal some myocardial structural and metabolic functions. A strong correlation between ongoing cardiac remodeling and the turnover of extracellular matrix proteins was reported in different studies [80,81]. Matrix metalloproteinases (MMPs) are the enzymes that degrade the extracellular matrix, increase matrix turnover, and change the expression of several micro-ribonucleic acids (mi-RNAs) that induce contractile dysfunction of the myocardium [72]. Elevated levels of MMPs, especially MMP-9, and reduced levels of the tissue inhibitors of MMPs are observed in myocardial fibrosis. The clinical usefulness of these new biomolecules for the diagnosis of DCM is currently under investigation.
Serum aminoterminal propeptide of type III, an indicator of type III collagen turnover in the body, was proposed as an early index of LV dysfunction in obese subjects with insulin resistance [82].
Recently, brain natriuretic peptide (BNP) emerged as a helpful biomarker for screening subclinical ventricular diastolic dysfunction in patients with uncontrolled diabetes [83,84]. Epshteyn et al. [85] showed a high positive predictive value of 96% for plasma BNP levels (> 90 pg/mL) in diabetic patients for the diagnosis of LV dysfunction with echocardiographic correlation.
Cardiac troponins (T, N, and I) are biomarkers released into circulation from the injured myocardium in inflammatory or ischemic disease. Elevated troponin T levels were found in infants with cardiac dysfunction and cardiomyopathy born to diabetic mothers [86]. However, the role of troponins for the assessment of adult patients with DCM is unclear.
Mi-RNAs are small non-coding RNA molecules that coordinate cellular gene expression. The dysregulation of mi-RNAs has been linked to diabetes and many of its complications. Altered levels of mi-RNAs were observed in the cardiomyocytes of experimental diabetic models [87]. These novel biomarkers may emerge as prognostic and diagnostic methods for patients with DCM in the future.
Detection of metabolic changes
A reduction in the phosphocreatine (PCr)/ATP ratio suggests suppressed ATP production and/or the suppressed production of PCr from ATP by the creatine kinase system. Determining PCr and ATP levels in the human myocardium by magnetic resonance spectroscopy (MRS) demonstrated that the PCr/ATP ratio is significantly reduced in diabetes and that the ratio is negatively correlated with plasma FFA levels or live triglyceride levels [77,88]. A promising novel approach for the diagnosis of DCM is the characterization of metabolic changes in the myocardium using 31P-MRS and 1H-MRS. The PCr/ATP ratio, an indicator of energy charge, is reduced in the myocardium of diabetic subjects compared with control patients. Recent studies using 1H-MRS showed that an increase in myocardial triglyceride content was associated with LV diastolic dysfunction in diabetic subjects [41,42].
PREVENTION AND THERAPEUTIC STRATEGIES
Recently, a better understanding of the pathophysiology and pathogenesis in patients with DCM has provided improved management options. These include lifestyle modifications, improved diabetic control, the management of coexistent hypertension and CAD if present, lipid-lowering therapies, and the management of heart failure (Table 2) [1].
Lifestyle modifications
Weight loss, limitation of fat and total energy intake, and regular physical activity can positively adjust metabolic abnormalities and improve tissue and systemic insulin resistance by increasing insulin-mediated glucose transport and post-receptor insulin signaling, which seems to be related to facilitated signal transduction at the level of phosphatidylinositol 3-kinase and insulin receptor substrate [56,67].
Physical activity was associated with a significant reduction in cardiovascular disease and all-cause mortality in patients with diabetes mellitus in many clinical studies [89]. Exercise training was beneficial for reducing the incidence of DCM in both human patients and animal models [83,84]. It is difficult to predict the benefit of physical activity in documented cases of the disease in the absence of controlled clinical trials. However, better diabetic control with regular exercise would have beneficial effects on disease outcome. Maintaining healthy eating patterns that are suitable for diabetic subjects can also be expected to offer similar beneficial effects [1].
Antidiabetic medications
Better glycemic control was associated with better outcomes in diabetic microcirculatory complications in many clinical trials. However, the beneficial effects of tight glycemic control on macrovascular outcomes are still unclear. Because microvascular disease plays an important pathogenic role in the development of DCM, rigid glycemic control would be expected to benefit patients [1].
Improved glycemic control delayed DCM in animal models [90]. Strict glycemic control improved stress-induced ventricular dysfunction without CAD in poorly controlled diabetic patients in a large prospective study [91]. Another case-controlled study using cardiac MRI in patients with T1DM demonstrated that rigorous glycemic control was associated with better DCM outcome parameters [92]. Diabetes management was also beneficial for reducing myocardial steatosis [93].
Metformin upregulates cardiac autophagy, which plays a role in the prevention of DCM in animal models [94]. Metformin was reported to reduce mortality and improve the clinical outcome in overweight patients with heart failure and diabetes mellitus, in spite of the increased risk of lactic acidosis [95]. This drug facilitates glucose uptake and GLUT4 translocation in insulin-resistant cardiomyocytes and the myocardium by activating 5´ adenosine monophosphate-activated protein kinase [95,96]. However, there are no data regarding its role in humans with DCM.
Thiazolidinediones, which are insulin sensitizers, improve contractile dysfunction and myocardial glucose uptake by activating PPAR-γ [97]. However, thiazolidinedione therapy can cause chronic symptoms that resemble heart failure by increasing water and sodium reabsorption in the kidney collecting tubules and vascular permeability, which induces generalized edema [98]. Therefore, the drug is generally not recommended in patients with heart failure. However, pioglitazone had anti-inflammatory effects that improved myocardial fibrosis in animal models, and so the drug may prevent the development of DCM [99].
Natural glucagon-like peptide 1 (GLP-1) has a very short biological half-life. Synthetic GLP-1 mimetic agents with longer half-lives, such as exenatide and liraglutide, are new anti-diabetic agents being widely used. Their use in obese T2DM patients is associated with significant improvements in weight loss and glycemic control. GLP-1 agonists also attenuate cardiomyocyte apoptosis in rat models [100]. GLP-1 encourages insulin secretion, enhances nitric oxide-induced vasodilation, and facilitates glucose use in the myocardium [97,101]. This novel group of drugs may emerge as a promising management strategy in obese T2DM patients with DCM.
Dipeptidyl peptidase 4 (DPP-4) is an enzyme that metabolizes endogenous GLP-1. DPP-4 inhibitors prolong the effects of endogenous GLP-1. Agents in this class, such as sitagliptin, linagliptin, saxagliptin, and vildagliptin, are effective anti-diabetic medications. They are weight-neutral and are especially useful in overweight and obese diabetic patients in combination with conventional anti-diabetic agents such as metformin. The use of sitagliptin promotes myocardial glucose uptake in patients with nonischemic cardiomyopathy [102]. DPP-4 inhibitors can prevent cardiac diastolic dysfunction and cardiac hypertrophy by inhibiting fibrosis and oxidative stress in mouse models of insulin resistance and obesity [103]. The possible therapeutic role of DPP-4 inhibitors in patients with DCM is yet to be elucidated.
Amylin analogues reduce HbA1c values, body weight, and even the insulin requirement when administered together with insulin [104]. Their role in controlling hyperglycemia may benefit patients with DCM.
Empagliflozin, an inhibitor of sodium-glucose cotransporter 2, is a new antidiabetic agent that reduces HbA1c levels in patients with T2DM by controlling visceral adiposity, blood pressure, arterial stiffness, albuminuria, weight, oxidative stress, hyperinsulinemia, and circulating uric acid levels [105]. In the Empagliflozin Cardiovascular Outcome Event Trial in Type 2 Diabetes Mellitus Patients (EMPA-REG OUTCOME) involving patients with T2DM, empagliflozin with standard therapy lowered the rate of primary composite cardiovascular outcome and death compared with placebo added to standard therapy; however, an increased rate of genital infections was noted [106].
Vasoactive medications and coronary intervention
There are no formal guidelines regarding the management of coexistent cardiac ischemia and hypertension in patients with DCM. However, when these diseases coexist they promote the progression of DCM because of their harmful effects on ventricular structure and function. The optimal treatment of CAD and hypertension would be expected to improve the disease progression and even slow it down. Coronary intervention in appropriate cases with significant CAD may improve the clinical outcomes and symptoms. The management of heart failure depends on the severity, type (diastolic or systolic), and associated conditions like CAD and hypertension [1].
Various vasoactive medications have been tried in both human patients and animal models with DCM, with variable results. The most-studied medications were those where subjects were active on the renin-angiotensin systems. The production of AGT II within the myocardium was proposed as a mechanism for the development of DCM. Recently, angiotensin converting enzyme inhibitor (ACEi), renin inhibitor (aliskiren), and angiotensin II receptor blocker (ARB) were all shown to be protective against DCM in rat models [107]. ARBs and ACEis were also beneficial in both human and animal models of DCM [108,109].
β-Adrenoreceptor blockers were effective in experimental models of DCM [110]. Because of the proven benefits of β-blockers in chronic heart failure, this group of drugs should be considered for treatment of DCM; however, there are no reported randomized clinical trials examining the benefits. They can be used as effective antihypertensive agents in DCM patients with hypertension.
Similarly, calcium channel antagonists were beneficial in animal models of DCM [111]. However, data on human subjects are lacking to make evidence-based recommendations for the use of these agents in the management of DCM, especially in the absence of coexistent hypertension.
Sildenafil, a selective phosphodiesterase type 5 inhibitor, was recently shown to improve myocardial function, cardiac remodeling, and some circulatory markers of cardiac inflammation in patients with DCM [112]. Larger clinical trials in the future may determine if this novel agent can be recommended for routine use in patients.
Lipid lowering medications
Dyslipidemias are more detrimental in diabetic than non-diabetic individuals because of their higher atherogenic potential. The particle size of low density lipoprotein cholesterol (LDL-C) is smaller in diabetic subjects, which is more atherogenic even with near-normal plasma levels. The use of statins reduces cardiovascular mortality and events in patients with diabetes and vascular risk factors in multiple clinical trials [113], and is beneficial even for primary prevention in patients without established cardiovascular disease [114]. The vascular remodeling capacity of statins is referred to as the pleiotropic effect.
Atorvastatin, reduces myocardial fibrosis, intramyocardial inflammation, and improves LV function in rat models of experimental DCM, independently of its LDL-C-lowering capacity [115]. Similarly, fluvastatin is useful for attenuating cardiac dysfunction and myocardial interstitial fibrosis in rat models of the disease [116]. Although there are no clinical trials investigating the role of lipid-lowering therapy in individuals with established DCM, the benefits of dyslipidemia treatment can be anticipated in these patients, along with a role in primary disease prevention.
Metabolic modulators
Trimetazidine, an atypical anti-anginal agent with antioxidant properties, is a competitive inhibitor of the terminal enzyme in β-oxidation; it might be used to improve substrate or myocardial metabolic flexibility and cardiac function in the setting of DCM [117]. Trimetazidine might affect myocardial substrate use by inhibiting oxidative phosphorylation and shifting energy production from FFAs to glucose oxidation [118]. It might also help reduce the injury caused by free radicals and calcium overload, preserve intracellular ATP and PCr levels, improve endothelial function, and inhibit cellular apoptosis [118]. The drug has promising beneficial effects on heart failure in diabetic patients with both idiopathic and ischemic dilated cardiomyopathy [119]. Animal models revealed that trimetazidine improved myocardial function by augmenting the oxidation status and attenuating lipotoxicity in the heart; therefore, it might suppress the development of DCM [120]. Human trials are needed to investigate the beneficial effects of this well-tolerated drug on the treatment and prevention of DCM.
Ranolazine, a potent late Na+ current inhibitor, might normalize altered cardiomyocyte intracellular Ca2+ concentrations due to the close relationship between Ca2+ and Na+ handling by the Na+/Ca2+ exchanger [121]. Ranolazine improved measures of hemodynamics, but not cardiac relaxation parameters [121]. A single treatment with ranolazine is probably not sufficient to affect cardiac function and structure [121].
Perhexiline is an inhibitor of carnitine 0-palmitoyltransferase 1; it increases LV ejection fraction, maximal O2 consumption (VO2 max), resting and peak stress myocardial function, and skeletal muscle energetics [117]. In patients with chronic heart failure, a multicenter double-blind randomized controlled trial showed that perhexiline improved LV ejection fraction, resting and peak stress myocardial function, VO2 max, and skeletal muscle energetics. Despite these benefits, the use of the perhexiline is diminishing due to reports of peripheral neuropathy and hepatotoxicity [122].
Although experimental models have shown that α-lipoic acid suppresses cardiac fibrosis and has beneficial effects on cardiac redox homeostasis [123], human data are not yet available. Other investigational antioxidants that were useful in recent animal models include resveratrol [124], luteolin [125], riboflavin [126], and sodium ferulate [127].
Resveratrol reduces blood glucose levels and improves cardiac function and insulin sensitivity in patients with heart failure [128]. Although resveratrol is associated with improved triglyceride levels, heart rate, and glycemia, it does not prevent cardiovascular- or cancer-, or all-cause mortality [129].
EMERGING TREATMENT MODALITIES
Targeting mitochondrial oxidative stress
Given the importance of oxidative stress in the development of DCM, targeting excess myocardial ROS production with novel antioxidants might offer a promising approach for preventing DCM (Table 3).
The Szeto-Schiller peptide peptide d-Arg-2′, 6′-dimethyltyrosine-Lys-Phe-NH2 (SS31) is a positively charged free-radical scavenger that can accumulate to high levels in the mitochondria and prevent diastolic dysfunction, fibrosis, and cardiac hypertrophy [101,130]. SS31 prevents cardiolipin from converting cytochrome into a peroxidase while protecting the cardiolipin electron-carrying function by interacting with cardiolipin [131]. SS31 also protects the structure of mitochondrial cristae and promotes oxidative phosphorylation [131]. Thus, SS31 represents a new class of compounds that can restore bioenergetics and recharge the cellular powerhouse.
Coenzyme Q10 improves cardiac function in patients with diabetes mellitus and concurrent heart failure [130,132]. A randomized controlled trial showed that long-term coenzyme Q10 treatment in patients with chronic heart failure is safe, improves symptoms, and reduces all-cause mortality by 42% and cardiovascular death by 43% [133].
Cell- and genetic-based therapy
Cell-based therapy and the genetic correction of abnormalities are potential strategies to prevent the development of DCM or treat the early stages of this condition [134].
For example, the gene delivery of nerve growth factor preserves microvessel density, cardiac perfusion, and LV diastolic and systolic function [135]. The systemic administration of Pim-1 improves LV diastolic function and prevents cardiac apoptosis, fibrosis, and the development of heart failure [136].
The dysregulation of mi-RNA function is an important pathogenic mechanism of diabetes and its complications such as DCM, and the artificial restoration of normal function can be a potential therapeutic target. Six mi-RNAs (miR-34b, miR-34c, miR-199b, miR-210, miR-223, and miR-650) might be involved in the pathogenesis of the failing myocardium in patients with diabetes mellitus [137]. Thus, mi-RNAs have become an active area of investigation to determine their potential contribution to heart disease in patients with T2DM. Mi-RNA-based therapies might be beneficial in patients with diabetes mellitus and cardiac pathology [137]. Specific mi-RNA targets are useful for the treatment of structural heart disease in mice [138]. Similarly, the transplantation of bone marrow-derived endothelial progenitor cells (stem cell therapy) ameliorates DCM in rat models [139]. Ongoing investigations may help us to translate the success of these experimental models into clinical practice in the future.
CONCLUSIONS
DCM is an important but lesser-elucidated complication of chronic diabetes, and is associated with significant cardiac mortality and morbidity. The wide discrepancy in the reported prevalence of DCM may be related to disparities in the types of diagnostic test used by various researchers. The pathophysiology and pathogenesis of DCM are still not fully understood, although proposed mechanisms include insulin resistance, microvascular disease, lipotoxicity, chronic hyperglycemia-associated metabolic and oxidative stress, coexistent CAD and hypertension, CAN, and altered immune responses. The manifestations of DCM can vary from subclinical ventricular dysfunction to overt heart failure. Echocardiography is currently the standard clinical diagnostic method for DCM. Newer investigative modalities such as cardiac MRI, PET imaging, radionuclide scans, and various serum/plasma markers are emerging as diagnostic tools. The management of DCM covers lifestyle modifications, medications for heart failure, good glycemic control, and the treatment of coexistent CAD, dyslipidemia, and hypertension. Novel therapeutic approaches including targeting mitochondrial oxidative stress and cell- or gene-based therapy are currently being investigated. Further research is needed to understand the exact mechanisms involved in the development and course of DCM to enhance the discovery of clinically effective targets for preventing this condition and its progression to heart failure.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print





