


INTRODUCTION
Rheumatoid arthritis (RA) is a chronic and debilitating autoimmune disease characterized by the interaction of various inflammatory mediators and cells [1]. Although the etiology of RA is not clear, inflammatory cytokines and tissue-destructive molecules play key roles in the initiation and progression of the inflammatory processes that characterize the disease [2]. Accordingly, biological agents that inhibit proinflammatory cytokines have been widely used in the treatment of RA [3].
Macrophage migration inhibitory factor (MIF) is an evolutionarily ancient and highly conserved cytokine that was originally described as an activity of cognate T cell supernatants that inhibits macrophage migration [4]. MIF is cloned and purified, and its activity characterized at the molecular level in 1993 [5]. MIF is structurally unique; it has a monomeric molecular mass of 12.5 kDa and consists of two antiparallel ╬▒-helices that together with six ╬▓-pleated sheets form the extended secondary structure of the molecule. Biophysical studies indicate that, in its active form, MIF is a homotrimeric molecule with topological homology to only one other mammalian protein, the enzyme d-dopachrome tautomerase [6], a broad-spectrum intra- and extracellular protein produced by a variety of cell types, including monocytes/macrophages [7], lymphocytes [8], eosinophils [9], neutrophils [10], epithelial cells [11,12], endothelial cells [13], and smooth muscle cells [14]. By contrast, MIF has a chemokine-like function, promoting the migration and recruitment of leukocytes to infectious and inflammatory sites [15].
MIF is stored in pre-formed cytoplasmic pools and is rapidly released in response to stimuli such as microbial products, proliferative signals, and hypoxia [7,16,17]. One of the earliest physiological functions described for MIF was as a counter-regulator of glucocorticoid-mediated suppression of immune cell responses [18], which is important for the regulation of systemic inflammatory responses in settings such as invasive stress or acute illness, characterized by high adrenal glucocorticoid levels. MIF also plays a pivotal upstream role in sustaining immune cell survival, by inhibiting activation-induced apoptosis. This effect serves to provide for optimal but, in some pathologic circumstances, excessive, inflammatory responses [19].
MIF participates in the pathogenesis of many inflammatory diseases, including colitis [20], multiple sclerosis [21,22], systemic lupus erythematosus [23,24], glomerulonephritis [25,26], psoriasis [27,28], and diseases more recently recognized as inflammatory, such as atheroma formation and even atheromatous plaque rupture.
MIF IN THE PATHOGENESIS OF RA
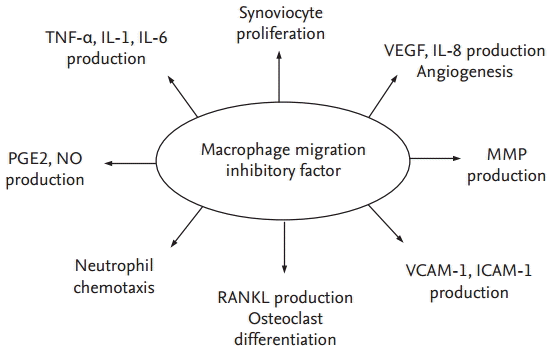
Expression of MIF is increased in the arthritic joints of patients with juvenile idiopathic arthritis and in those with RA [29-31]. Serum and synovial fluid levels of MIF are also higher in RA patients than in either osteoarthritis patients or healthy volunteers. These high levels are associated with bone erosion and disease activity [26,32-34].
MIF stimulates the release of tumor necrosis factor (TNF), interleukin IL-1, IL-6, IL-8, and prostaglandin from macrophages and synovial fibroblasts to induce matrix metalloproteinase (MMP)-1 and MMP-3, phospholipase A2, and cyclooxygenase-2, which together lead to tissue degradation in RA-related processes [35]. MIF also induces MMP-9 and MMP-13 in rat osteoblasts, which may be relevant to the bone destruction and osteoporosis characteristic of RA [36]. Tissue degradation by MMPs is a typical pathological feature of RA. MIF contributes to this process by up-regulating MMP-1 and MMP-3 mRNA levels in synovial fibroblasts, which in large part are responsible for the degradation of extracellular matrix components in RA (Fig. 1) [35]. The up-regulation of MMP genes by MIF is probably mediated by a complex regulatory system. Glucocorticoids repress MMP-1 gene transcription via the interaction of glucocorticoid receptor proteins with the activator protein (AP)-1 complex [37]. The interaction of MIF and glucocorticoid/AP-1 was reported by Chauchereau and coworkers [38], who showed that Jun activation domain-binding protein-1 (Jab1)/CSN5 COP9 signalosome subunit 5 (CSN5) binds to the glucocorticoid receptor. Together, these studies demonstrated the potent counter-regulatory activity of MIF in the glucocorticoid-mediated control of inflammation in general [39] and synovial inflammation in particular [40].
MIF induces proliferation of human RA synovial fibroblasts and inhibits both p53 expression and apoptosis [41,42]. Accordingly, MIF inhibition would convey a proapoptotic signal to these hyperplastic cells. MIF-mediated inhibition of p53 expression and apoptosis prolongs cell survival and has been described in macrophages [19]. MIF also interacts closely with various proinflammatory cytokines and plays a major role in innate immunity against bacterial infections, by enhancing TNF-╬▒ secretion [5], Toll-like receptor 4 expression [43], phagocytosis, and intracellular killing mechanisms [44]. In addition, it is equally efficiently involved in the adaptive immune response, by favoring Th1 activation and differentiation. The autocrine and paracrine effects of MIF on immune cell activation include the induction of IL-1 and TNF, which in turn stimulate further MIF production [18,45]. Endogenous MIF is required for IL-1- and TNF-induced mitogen-activated protein kinase (MAPK) activation and regulates the expression of the receptors for these cytokines [46].
Anti-TNF therapy in RA patients diminishes the serum levels of the chemokine chemerin, a specific chemoattractant for macrophages and dendritic cells. The suppression of these MIF-producing cells decreases serum MIF concentrations, which in turn reduces inflammation [47]. The arthritic joints in MIF-mice have lower serum IL-1╬▓ and IL-6 levels, but the levels of these cytokines are restored when the defect in these mice is reconstituted using wild-type macrophages.
Vascular endothelial cells play vital roles in systemic inflammatory and immunological events; for example, the cytokines and growth factors produced by endothelial cells participate in complex inflammatory processes [48]. MIF mRNA is not limited to T lymphocytes or macrophages but is also expressed in vascular endothelial cells. In RA patients, serum and synovial fluid MIF levels correlate with vascular endothelial growth factor (VEGF) levels. MIF stimulates synovial fibroblasts to produce VEGF and IL-8; in vitro, it stimulates human umbilical vein endothelial cells to increase vascular tube formation [49]. Endothelial cells also readily secrete their cytoplasmic stores of MIF in response to the presence of a bacterial component, such as in infectious states. MIF subsequently initiates production of the proinflammatory cytokines TNF-╬▒ and IL-1. Thus, MIF is not only an initiator of the inflammatory process involving endothelial cells but also acts as a growth factor in the response to cellular damage.
Although the data are controversial, an osteoclastogenic role for MIF in human RA has been reported. MIF stimulates both the production of receptor activator of nuclear factor kB ligand (RANKL) by RA synovial fibroblasts and the differentiation of peripheral blood monocytes to mature osteoclasts [50]. MIF-triggered RANKL expression is partially reduced by blockage of IL-1╬▓, while osteoclastogenesis is suppressed by inhibition of nuclear factor-╬║B (NF-╬║B), phosphatidylinositol 3-kinase (PI3K), p38 MAPK, and AP-1. Other studies have also shown that MIF significantly up-regulates MMP-13 mRNA expression in rat primary osteoblasts, by activating Src-related tyrosine kinase, extracellular signal regulated kinase (ERK) 1/2, and AP-1-dependent signaling pathways [36]. Collectively, these results support a role for MIF as an upstream regulator of synovial cytokine expression in RA.
An association of MIF polymorphisms with RA has been described in a number of studies. In their study of a Chinese population, Liu et al. [51] reported that the MIF-173 C allele, in which there is an alteration in the MIF promoter region, may contribute to RA susceptibility and increase the risk of RA. Similarly, an association between the ŌłÆ794 CATT7 and ŌłÆ173*C alleles, which are in linkage disequilibrium, and the high clinical activity of RA has been reported [52]. Thus, MIF polymorphisms may be associated with both a higher risk and a greater severity of RA.
Intracellular signal pathways of MIF
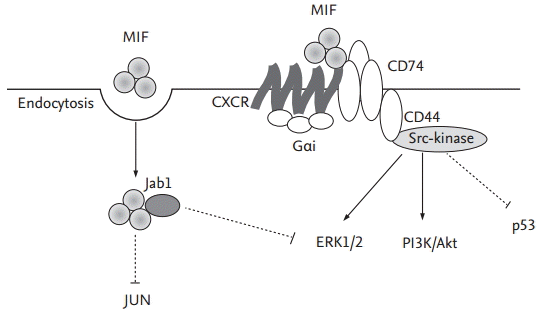
The signal transduction pathways used by MIF in its activation of cells and cellular events is incompletely defined, although cell-surface-receptor-mediated pathways have been implicated (Fig. 2) [53]. Recently, CD74, the cell surface form of the class II invariant chain, was identified as the receptor for MIF [54]. The interaction of MIF with CD74 activates MAPK pathways [53], and the phosphorylation of MAPKs leads to the expression of target genes that are important in inflammation and proliferation. These observations highlight the importance of MAPKs in RA [55]. The activation of distinct MAPK subtype cascades is dependent on the cell type and the nature of the stimuli used; furthermore, the functional role of each MAPK may differ depending on the cell type. In general, the ERK cascade mediates the proliferation, differentiation, and survival of signal-promoting cells, whereas both p38 and JNK MAPKs are involved in cell responses to environmental stresses and inflammatory cytokines [55,56]. MIF-induced ERK activation is associated with cell proliferation and prostaglandin E2 production [53]. This is in keeping with the induction by MIF of a uniquely sustained phosphorylation of ERK [19], associated with increased NIH/3T3 proliferation and enhanced phospholipase A2 activity. The finding that MIF up-regulation of these cellular events is not accompanied by p38 phosphorylation suggests the nonutility of this pathway. Although we demonstrated that MIF induces phosphorylation of p38 MAPK in RA synovial fibroblasts [57], the MIF-induced activation and proliferation of synovial fibroblast is mainly mediated by the ERK pathway [42,57]. Similarly, MIF phosphorylation of the ERK pathway occurs in the up-regulation of N-Myc protein expression in neuroblastoma tissues [58], in the growth and angiogenesis of murine colon cancer cells [59], and in the up-regulation of MMP-1 in human dermal fibroblasts [60]. The latter study also shows that MIF phosphorylation of the JNK but not the p38 signaling pathway is involved in stimulating MMP-1 expression [60]. The MIF-mediated activation of the MAPK pathway is further confirmed in a report showing the activation by MIF of the c-jun element of AP-1 transcription factor in its stimulation of MMP-1 and -3 expression [35,36]. In addition, MIF activates PI3K and its effector kinase, Akt, to promote tumor growth and angiogenesis [59,61]. Clearly, MIF differentially activates distinct signaling pathways to stimulate target cells and cellular events.
The intracellular actions of MIF have also been studied. At high concentrations MIF influences the transcriptional activity of AP-1, by interacting with Jab1 [62]. This interaction interrupts AP-1-dependent gene transcription and inhibits the growth-promoting effects of Jab1 on fibroblasts. These events demonstrate the contrasting effects of MIF on inflammation and proliferation, which may be related to its relative concentration. Thus, high concentrations of MIF may inhibit AP-1-dependent events to prevent over-activation of the immune response [53,63,64].
THERAPEUTIC EFFECTS OF MIF INHIBITION IN RA ANIMAL MODELS
A role for MIF in inflammatory joint disease is first explored in the collagen-induced arthritis (CIA) mouse model, which showed that MIF antagonism delays the onset and decreases the frequency of arthritis [65]. MIF promotes Th1 immunity and anti-MIF treatment lowers serum immunoglobulin G2a levels, without significant effects on collagen type II-induced interferon-╬│ production. Moreover, the overall T-cell proliferative response to collagen type II is surprisingly higher in anti-MIF-treated mice [65]. Further evidence of a role for MIF in RA comes from two cell-mediated animal models of RA. In rat adjuvant arthritis (AA), anti-MIF therapy dose-dependently reduces disease severity [66]. During disease development, MIF levels are increased in sera and synovial tissue, and an association between synovial MIF and ED1-positive macrophages has been reported [66]. Similarly, MIF antagonism decreases the severity of antigen induced arthritis (AIA) in mice, as measured by synovial hypercellularity, and glucocorticoid treatment impedes disease development [40]. Glucocorticoid regulation of MIF is confirmed in vivo in AA rats [67]. Adrenalectomy prior to AA induction results in increased joint inflammation; in these animals, serum and pituitary MIF levels are increased but, surprisingly, the levels in the synovium are decreased. Nonetheless, MIF regulation of joint inflammation is still significant, as the protective effects of anti-MIF treatment are retained [67]. These findings suggest differential regulation of local and systemic MIF in the context of AA.
Further support for the role of MIF in RA comes from MifŌĆō/ŌĆō mice. Two studies demonstrate suppression of CIA in MifŌĆō/ŌĆō mice [68]. In the AIA model, MifŌĆō/ŌĆō mice has a reduced severity of histological arthritis, including evidence of reduced cartilage damage [41]. The latter study also shows reduced proliferation of synoviocytes as well as increases in p53 expression and apoptosis in these cells in the absence of MIF (synoviocyte expansion contributes significantly to the development of joint damage in RA by facilitating the invasion of synovium into cartilage and bone). Studies using MifŌĆō/ŌĆō mice also implicates MIF in the regulation of leukocyte recruitment in response to stimuli such as endotoxin and TNF, and directly demonstrated a requirement for MIF in leukocyte recruitment into the joint [69]. These observations suggest that MIF contributes to the hypercellularity of RA synovial lesions through its effects on leukocyte recruitment, proliferation, and survival (Table 1).
MIF may also play a role in the blunted response to steroids. In the study by Santos and coworkers [40], dexamethasone treatment induces inhibition of AIA, whereas MIF treatment reverses the effect of the administered steroid. AIA is significantly inhibited by anti-MIF monoclonal antibodies whereas the synthesis of MIF by synovial cells is enhanced by low concentrations of glucocorticoids.
MIF is a proinflammatory cytokine with a broad range of cellular targets and functions. Other soluble proinflammatory cytokines, such as TNF and IL-1, have been successfully targeted in RA and other inflammatory diseases using bioengineered soluble receptors or receptor antagonists and specific antibodies [70-73]. As a soluble cytokine, MIF and its recently discovered cell surface receptor CD74 suggest the potential of current technologies in targeting MIF in human inflammatory diseases. More importantly, the unique glucocorticoid-antagonistic capability of MIF provides an additional potential target in patients who have become resistant to glucocorticoid therapy during treatment for autoimmune disease. Continued investigation of the molecular immunology of MIF will provide better strategies to target it therapeutically. The success of this approach in RA will include reductions in inflammation, the protection of cartilage and bone, and the favorable reversal of the deficient apoptosis of RA synoviocytes, while leaving NF-╬║B dependent host defenses intact.
Taken together, these studies recommend further studies of MIF as a potential therapeutic target for RA. However, these must be preceded by elucidation of the role of MIF in RA.
CONCLUSIONS
The various roles of MIF in the pathogenesis of RA include its promotion of the synthesis of proinflammatory cytokines and tissue-degrading molecules as well as induction of osteoclast differentiation. The inhibition of MIF in animal models of arthritis is proof of the efficient therapeutic effect of this approach in blocking the initiation and progression of arthritis. Small molecular inhibitors that regulate MIF or its signaling pathways may provide new therapeutic options for managing RA patients.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print





