


INTRODUCTION
Asthma is a chronic airway inflammatory disease characterized by airflow obstruction and bronchial hyperresponsiveness [1,2]. Asthmatic airway inflammation has heterogenous clinical phenotypes which can depend on different cellular inflammatory pathogenic mechanisms [3]. Eosinophil is considered as a key effector cell in asthma [4,5]; however, abundant published data reported strong associations of airway neutrophilic inflammation with severe asthma, corticosteroid treatment responses, asthma exacerbations and asthma phenotypes/endotypes [6]. The roles of neutrophil in asthma pathogenesis are not completely understood and the genetic predictive factor for neutrophilic inflammatory phenotypes in asthma has not been identified.
Macroautophagy (referred as autophagy in this report) is a cellular process important to cell survival, by which damaged organelles and proteins are sequestered to autophagosomes and delivered to lysosomes for degradation [7]. Among the proteins that take part in autophagy pathway, autophagy related (ATG) 5 protein in the conjugation with ATG12, and ATG7 acting as E1-like enzyme play important roles in the maturation of autophagosomes by facilitating the elongation of isolation membranes (phagophore). Recently, autophagy has been implicated in inflammation and immune responses of airway diseases such as asthma and chronic obstructive pulmonary disease [8]. Autophagy plays crucial roles in innate as well as adaptive immune responses by supporting the proliferation, activation and function of various immune cells such as macrophages, neutrophils, mast cells, T and B cells [9-11]. Regarding neutrophils, autophagy was found to regulate cell differentiation, formation of neutrophil extracellular trap (NET) and cell death [12], which could contribute to the pathogenesis of immune diseases such as asthma. In asthma patients autophagosomes were detected more frequently in epithelial cells compared to those in healthy subjects [13]. In addition, single nucleotide polymorphisms (SNPs) of ATG5 such as rs510432 and rs12212740 were found to associate with asthma susceptibility and lung function [13,14]. These findings suggest an association of autophagy and ATG genetic polymorphisms in asthma pathogenesis.
Based on the findings, we investigated the association of ATG5 and ATG7 genetic polymorphisms with asthma susceptibility, severity and clinical features with a focus on neutrophilic inflammation in the present study.
METHODS
Study subjects recruitment
We enrolled 408 asthmatic patients and 201 healthy normal controls (NCs) from Ajou University Hospital (Suwon, Korea) to a case-controlled study. Asthma was diagnosed at the first evaluation based on a history of respiratory symptoms as well as the results of airway reversibility and hyperresponsiveness to methacholine following the Global Initiative for Asthma guideline. Severe asthma was defined as asthma that requires treatment with high dose inhaled corticosteroids plus a second controller and/or systemic corticosteroids to control it or that remains “uncontrolled” despite this therapy, follows American Thoracic Society guidelines [15]. All asthma patients were recruited when the disease was stable on their regular medications without any viral or bacterial airway infection. NCs were healthy individuals who had no history of asthma symptoms. All the subjects were provided written informed consents prior to participating in this study.
Atopy was defined as one or more positive reactions on a skin prick test with 55 common inhalant allergens (Bencard Co., Brentford, UK) with histamine and saline controls. Methacholine bronchial challenge tests were performed as previously described using doubled doses of methacholine (0.075 to 25 mg/mL) [16]. The methacholine PC20 value (the concentration of methacholine needed to produce a 20% decrease in forced expiratory volume in 1 second [FEV1]) was determined by interpolation from a dose-response curve.
Sputum induction and blood collection
Sputum induction was performed as previously described [17]. Briefly, asthma subjects were pretreated with 200 μg salbutamol through a metered dose inhaler. The subjects then were inhaled nebulized sterile 3% saline solution for 20 minutes through an ultrasonic nebulizer (Omron Co., Kyoto, Japan). Expectorated sputum was collected into a petri dish after excluding the saliva. Simultaneously with sputum collection, venous blood was collected into acid citric dextrose containing tubes (BD Falcon, Franklin Lakes, NJ, USA) for genomic DNA preparation and Vacuette tubes (Greiner Bio-One, Monroe, NC, USA) for serum collection.
Assessment of sputum neutrophil count
Each sputum sample in petri dish was weighted and transferred into a 50 mL polystyrene tube. Four times volume (v/w) of freshly prepared dithiothreitol 0.1% (DTT, Sigma, St. Louis, MO, USA) diluted in distilled water was added to each sputum tube followed by incubation at 37℃ for 20 minutes with occasionally gentle vortex to dissociate the disulfide bonds of the mucus. The reaction was stopped by added phosphate buffer saline (PBS) in a volume equal to the sputum plus DTT solution. The tubes were centrifuged at 1,500 rpm for 5 minutes. The cell pellet was resuspended in 50 mL PBS and filtered through a 40 μm nylon filter (Millipore, Bedford, MS, USA). Total cell count and cell viability were determined by staining with Trypan Blue (Sigma) and observing under an inverted microscope. To assess differential cell count, a cytospin slide of each sample was prepared, fixed with methanol and performed hematoxylin and eosin stain. All slides were overread (total 500 cells) and the samples with more than 80% squamous cells were excluded from analysis. Sputum neutrophil count was expressed as percentage of neutrophils among non-squamous cells in each sample.
SNP selection and genotyping
Genomic DNA was prepared from peripheral blood samples using Puregene Blood Core Kit (Qiagen, Germantown, MD, USA) following the manufacturer’s protocol. We screened for SNPs located on promoter and 5’-untraslated region (UTR) of ATG5 and ATG7 from 40 healthy Korean volunteers by direct sequencing. Three SNPs of ATG5 including –769T>C, –335G>A in promoter region and 8830C>T in 5’UTR, were selected. Two SNPs of ATG7, including –100 A>G in promoter and 25108G>C in 5’UTR, were selected. ATG5 and ATG7 are located on chromosome 6 and 3, respectively. The information of ATG5 and ATG7 polymorphisms are shown in Table 1. Minor allele frequency of all selected SNPs was over than 5%. Target SNPs were genotypes from all genomic DNA samples of study subjects using a primer extension method and the SNaPshot ddNTP primer extension kit (Applied Biosystems, Foster City, CA, USA) with primers shown in Table 2.
Preparation of ATG5 constructs for promoter assays
A 3201 base-pair DNA fragment that contains ATG5 –769T>C and –335G>A was amplified using the following primers: forward primer 5’-CTCTATCGATAGGTACCAAGTGATGGTTAGGGTTCATGG-3’ and reverse primer 5’-GATCGCAGATCTCGAGGCCCTCCGTGTTCTGCCTAA-3’. The 3’ end of each primer (underlined) contains homologous sequences with ATG5 and the 5’ ends contain 15-16 bases homologous to each end of the linear pGL3-basic vector (Promega, Madison, WI, USA). Linear vector was prepared using Kpn1 and Xho1 restriction enzymes (Takara, Shuzo, Japan). The PCR amplified fragments were gel-purified using Expin Gel kit (GeneAll, Seoul, Korea) and then cloned into the linear pGL3-basic vectors using In-fusion HD cloning kit (Clontech Laboratories Inc., Mountain View, CA, USA) following the manufacturer’s manual.
Cell culture
Human airway epithelial cell (A549) and human mast cell (HMC-1) were purchased from American Type Culture Collection (Manassas, VA, USA). A549 cells were cultured in RPMI-1640 and HMC-1 cells were cultured in Iscove’s modified Dulbecco’s medium (Gibco, Grand Island, NY, USA) supplemented with 10% heat-inactivated fetal bovine serum (FBS), 100 U/mL penicillin G sodium and 100 μg/mL of streptomycin sulfate (Gibco). The cells were maintained at 37oC in 5% CO2 humidified atmosphere.
Endogenous mRNA expressions of ATG5 and ATG7
Total mRNA was extracted from various cell lines using Easy-Blue reagent (Intron Biotechnology, Seongnam, Korea) according to the manufacturer’s protocol. cDNA was synthesized by reverse transcription (RT)-PCR from 3 μg of total mRNA using MMLV-RTase (Promega) under optimized reaction condition (reaction buffer: 50 mM Tris-HCl pH 8.3, 17 mM KCl, 3 mM MgCl2, 10 mM DTT, 2 mM dNTP, 0.5 U/mL RNase inhibitors [Promega] at 42˚C for 1 hour 10 minutes and extanded at 70˚C for 10 minutes). The mRNA expression levels of ATG5 and ATG7 was evaluated by PCR using the specific primers (ATG5: forward 5’-TCTCTTGACTGCAGTACCTTTTC-3’ and reverse 5’-CGTGTCTGATACTGTAAGCCTTT-3’; ATG7: forward 5’-CGACGCTCAGTAGCCTGTAG-3’ and reverse 5’-TTTCTATCCCCTCCCCCTGG-3’), which was normalized by beta-actin mRNA expression level using specific primers (forward 5’-TCCTTCTGCATCCTGTCGGC-3’ and reverse 5’-CAAGAGATGGCCACGGCTGC-3’).
Transient transfection and dual luciferase assay
A549 cells were transfected with constructed plasmid DNA using lipofectamine (Invitrogen, Carlsbad, CA, USA) following the manufacturer’s protocol. Briefly, 1 × 105 cells/well were seeded onto 12-well plate to obtain 70% to 80% confluence. Cells were transfected with 1 μg of constructed plasmid DNA, 5 ng of Renilla plasmid DNA, 5 μL lipofectamine and grown for 48 hours. HMC-1 cells were transfected with plasmid constructs using a microporator (MP-100, Digital Bio Technology, Seoul, Korea) and Neon transfection system (Invitrogen). Each well of 12-well plate was filled with 1 mL of appropriate medium supplemented with 10% FBS. For each well, 5 × 105 cells mixed with constructed plasmid DNA (1 μg) and Renilla plasmid DNA (5 ng) were suspended in 10 μL of solution R. The mixture then was shocked at 1,650 V/20 mA/plus width 1 in the microporator pipette station containing 3 mL of solution E. Cells were then transferred into 12-well plate and incubated for 24 hours at 37˚C in 5% CO2 humidified atmosphere. Transfected cells were lysed with passive lysis buffer (Promega). 10 μL of each lysate was applied to a dual-luciferase reporter assay (Promega) using a Clarity Luminescence microplate reader (BioTek, Winooski, VT, USA) following the manufacturer’s protocol. The pGL3-control (Promega) and promoter-less pGL3-basic vectors were used as positive and negative control, respectively. Transfection and luciferase assays were replicated in three independent experiments.
Measurement of serum interleukin 8 by ELISA
Levels of serum interleukin 8 (IL-8) in were measured by using an enzyme-linked immunosorbent assay (ELISA) kit (Endogen, Woburn, MA, USA) according to the manufacturer’s protocol.
Statistical analysis
The chi-square test with one degree of freedom was used to test the frequency of each SNP for significant departure from the Hardy-Weinberg equilibrium in NC group; and p values of less than 0.01 were considered statistically significant [18]. The haplotype block pattern was constructed using Haploview version 4.2 (http://haploview.software.informer.com/4.2/). Haplotypes that had frequency of more than 5% were selected for statistical analysis.
Clinical characteristics were compared between the two study groups using Student t test and chi-square test. Differences in genotype and haplotype frequencies between the study groups were examined by a logistic regression analysis with codominant, dominant and recessive models after accounting for age and sex as covariates. Generalized linear models were applied to compare sputum neutrophil count and serum IL-8 level according to genotypes and haplotypes distribution in the asthma group. The statistical analyses used IBM SPSS version 20 (IBM Co., Armonk, NY, USA) and p values of less than 0.05 were considered statistically significant.
RESULTS
Clinical characteristics of study subjects
Table 3 demonstrates the clinical characteristics of study subjects. Asthma patients were older than NCs (42.4 ± 13.3 vs. 28.6 ± 12.3, p < 0.001). Smoking and atopy rate were significantly higher in the asthma group compared to those of NC group (p = 0.003 and p < 0.001, respectively). Among asthma patients, 17.3% were classified as severe asthma. Mean sputum neutrophil count of the asthma group was 53.6% ± 34.8%.
Allele, genotype and haplotype frequency of ATG5 and ATG7 polymorphisms
The distributions of the three SNPs of ATG5 and two SNPs of ATG7 did not depart significantly from the Hardy-Weinberg equilibrium in NC group (p > 0.01 for all SNPs) (Table 1). All the SNPs of each ATG5 and ATG7 were in strong linkage disequilibrium (Fig. 1). Interestingly, the two promoter SNPs of ATG5 gene (–769T>C and –335G>A) were in a complete linkage disequilibrium in our study population; consequently, the statistically analyzed data of –335G>A were shown as the representatives for the two ATG5 promoter SNPs. Three common haplotypes (HT1 [AC], HT2 [GT], HT3 [GC]) for ATG5 –335G>A and 8830C>T; and two common haplotypes (HT1 [AG], HT2 [GC]) for the two SNPs of ATG7 were constructed using Haploview software. The haplotypes that have a frequency more than 5% were selected for analysis. The distribution of genotypes and haplotypes of ATG5 and ATG7 polymorphisms in the two study groups was not significantly different (Tables 4 and 5); and no significant differences in those distributions were noted according to severity of asthma (Tables 6 and 7).
Clinical characteristics according to ATG5 and ATG7 polymorphisms
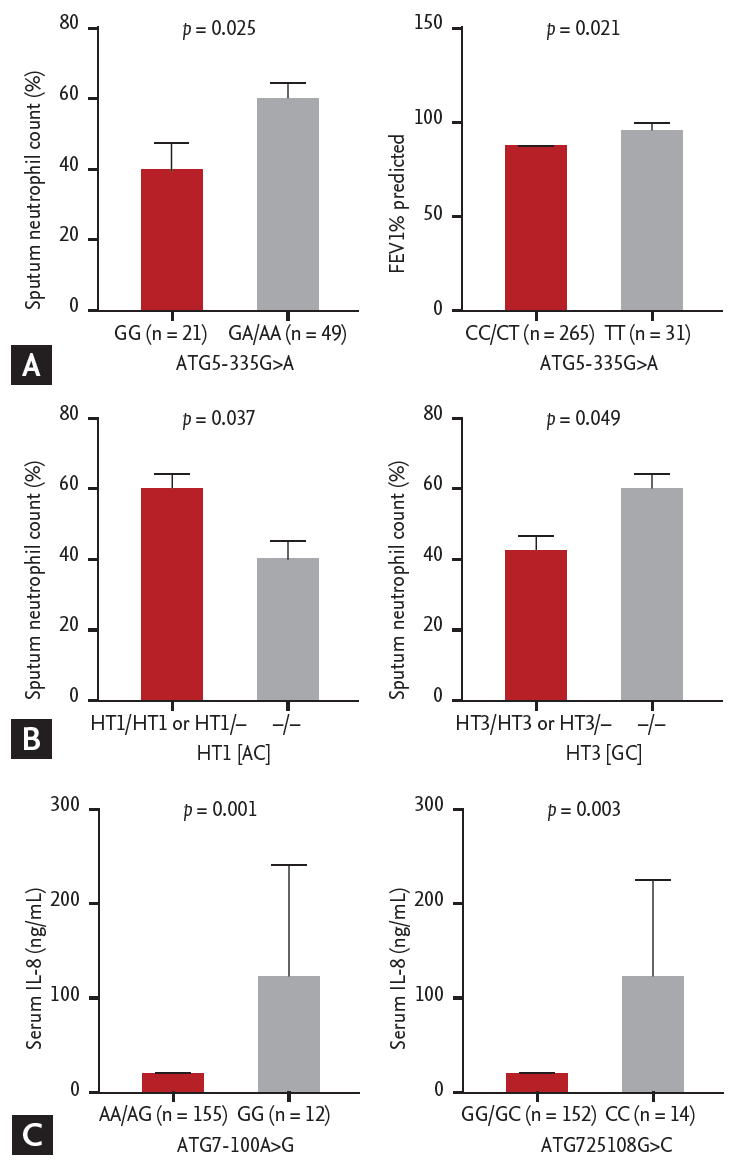
Fig. 2A shows the comparison of clinical characteristics according to the distributions of ATG5 genotypes in the asthma group. Asthmatics that carried GA/AA genotype at ATG5 –335G>A had significantly higher sputum neutrophil counts compared to those carrying GG genotypes (59.5% ± 33.5% vs. 39.8% ± 34.9%, p = 0.025). Asthmatics carrying CC/CT genotype at ATG5 8830C>T had significantly lower FEV1% predicted values compared to the carriers of TT genotype (83.3% ± 22.1% vs. 92.3% ± 19.2%, p = 0.021). Additionally, the prevalence of asthmatics with FEV1% predicted value < 80% tended to be higher in those carrying ATG5 8830 CC/CT genotype than in those carrying TT genotype (41.0% vs. 22.6%, p = 0.053). Fig. 2B shows that asthmatics carrying ATG5 HT1 [AC] haplotype had a significantly higher sputum neutrophil count compared to non-carriers (58.9% ± 33.6% vs. 39.6% ± 35.5%, p = 0.037); while asthmatic carriers of HT3 [GC] haplotype had a slightly but significantly lower sputum neutrophil count compared to non-carriers (43.5% ± 35.1% vs. 60.1% ± 33.6%, p = 0.049). No associations of HT2 [GT] with clinical parameters were noted.
Regarding ATG7 polymorphisms (Fig. 2C), serum IL-8 levels were significantly higher in asthmatic carriers of GG genotype at –100A>G as well as those carrying CC genotype at 25108G>C compared to the carriers of AA/AG and GG/GC genotypes (137 ± 445 pg/mL vs. 24.2 ± 55.5 pg/mL, p = 0.001; 123 ± 418 pg/mL vs. 24.5 ± 55.9 pg/mL, p = 0.003; respectively).
Transcriptional activity of ATG5 promoter polymorphisms
In order to select the target cell lines to investigate the functional promoter activity of ATG5 promoter polymorphisms, we evaluated the endogenous expression of ATG5 in various human cell lines including lung epithelial cell (A549), bronchial epithelial cell (Beas-2B), promyelocytic leukemia cell (HL-60), leukemic monocyte lymphoma cell (U937) and mast cell (HMC-1). A549 and HMC-1 cell line, known to play role in asthma pathogenesis, were selected based on their high expressions of ATG5 (data not shown).
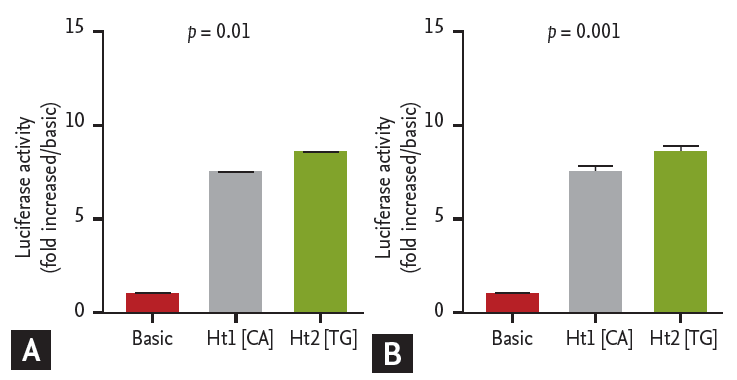
We constructed two DNA fragments that contained Ht1 [CA] with two minor alleles, and Ht2 [TG] with two major alleles of ATG5 promoter SNPs as described in the Method section for dual luciferase assays. Plasmids with DNA fragments containing major haplotype exhibited significantly higher luciferase activity compared to those containing minor haplotype in both A549 (p < 0.01) (Fig. 3A) and HMC-1 cells (p < 0.001) (Fig. 3B).
DISCUSSION
Previous reports demonstrated the associations of ATG5, but not ATG7, polymorphisms with asthma susceptibility and lung function [13,14]. In the present study with Korean adult asthmatics, we identified two promoter SNPs of ATG5, –769T>C and –335G>A, associated with sputum neutrophil count; and two SNPs of ATG7 gene, –100A>G and 25108G>A, associated with serum level of IL-8.
In a previous genome wide association study with White/Caucasian children asthmatics, allele G of ATG5 –335G>A (rs510432) was found to be associated with increased asthma risk. In the present study, the association of ATG5 –335G>A with asthma susceptibility was not noted; however, GA/AA genotypes at ATG5 –335G>A was associated with a higher sputum neutrophil count in adult asthmatic patients. Intriguingly, the two ATG5 promoter SNPs were in a complete linkage equilibrium in our study population, which explains the same finding observed in regards to the sputum neutrophil count in association with ATG5 –769T>C. These findings suggested a possibly functional effect of the two ATG5 promoter SNPs on transcriptional activity.
Martin et al. [14] demonstrated that ATG5 –335G allele increased a higher promoter activity compared to A allele; and two asthma-associated transcription factors, STAT1 and C-Fos, were assumed to bind to ATG5 –335G>A. Consistently, we found that Ht1 [TG] of ATG5 –769T>C and –335G>A enhanced significantly higher promoter activity compared to Ht2 [CA]. We do not have a direct evidence to explain for that the major haplotype of ATG5 promoter polymorphisms was associated with lower sputum neutrophil counts but enhanced the higher promoter activity. However, Yousefi et al. [19] identified a calpain-dependently cleaved fragment of mouse ATG5 protein related to spontaneous neutrophil apoptosis, which was independent to autophagy process. In addition, ATG5 could contribute to cell death by interaction with Fas-associated protein with death domain under the stimulation of interferon gamma (INF-γ) [20]. Because the formation of autophagosome in mouse only needs a small amount of ATG5 [21], the overexpression of ATG5 in human, which is affected by ATG5 promoter polymorphisms, probably increases intracellular autophagy levels and/or elicits neutrophil apoptosis mediated by the ATG5 cleaved fragment.
Asthmatics carrying C allele of ATG5 8830C>T had a lower FEV1% predicted value than carriers of the TT genotype. However, we did not observe a difference in the transcriptional activity of the two alleles of this SNP (data not shown). In the present study population, we observed that all subjects carrying ATG5 –335A allele concomitantly carried ATG5 8830C allele; however the reverse linkage was not found. This linkage explained for that HT1 [AC] and HT3 [GC] of ATG5 –335G>A and 8830C>T were associated only with sputum neutrophil count, but not FEV1% predicted value. This finding suggests that the two SNPs of ATG5 could play role in asthma pathogenesis by different mechanisms, which needs to be revealed by further studies.
GG genotype at ATG7 –100A>G and CC genotype at ATG7 25108G>C was also found to be associated with high serum levels of IL-8, a major chemoattractant and stimulator of neutrophils. However, these two variants did not exhibit different transcriptional activities in our transfection and luciferase assay system (data not shown).
The fact that the two promoter SNPs of ATG5 and the two SNPs of ATG7 were all associated with neutrophilic inflammatory markers suggested an interaction between the two genes. However, we did not find the associations of ATG5 and ATG7 polymorphisms in genegene interactions with asthma susceptibility as well as asthma clinical parameters in the present study (data not shown). This finding suggests independent roles of the two genes in neutrophilic inflammation of asthma.
Autophagy machinery was identified in murine as well as human neutrophils [22,23]. Reactive oxygen species (ROS) possibly trigger and regulate autophagy in neutrophils and other cell types [23,24]. Inflammatory cells including neutrophils that are recruited into the asthmatic airways produced greater ROS levels compared to those in normal subjects [25]. Concomitantly, greater number of double membrane autophagosomes were depicted in airway epithelial cells and fibroblast from asthmatics compared to those from healthy subjects [13]. These findings suggested a high level of autophagy occurring in cells that are involved in asthma pathogenesis, including neutrophils.
It is not fully understood if autophagy is a harmful or protective mechanism in asthma. However, autophagy has been known to protect the cells from damage or even cell death, as well as be essential for the survival, differentiation and function of various kinds of cell involved in inflammatory and immune responses [10,26-28]. Consequently, autophagy is able to augment the effects of immune cells to prolong airway inflammation that increase asthma severity. In addition, respiratory viral infection, which could lead to asthma exacerbation, also could activate ATG genes and autophagy [29]. Concurrently, Remijsen et al. [12] demonstrated that autophagy is crucial for the formation of NET which was identified in airways of allergic asthma patients and could be triggered by IL-8 [30,31]. NET is a structure composited by DNA net with histones and coated by several proteins such as neutrophil elastase and myeloperoxidase released from activated neutrophils. The role of NET in asthma pathogenesis is under investigations; however, neutrophil enzymes that coat the NET may damage the airway epithelia [32]. IL-8 levels, sputum neutrophil counts and NETs are all increased in neutrophilic asthmatics, and likely contribute to asthma severity [32-35]. Additionally, both neutrophils and IL-8 are features of difficult-to-treat asthma [36]. These findings suggest a potent role of autophagy in neutrophil activation which may enhance the severity of asthma.
We had a limitation in investigating the gene-environment interaction in the present study, which could be strictly evaluated by a further prospective study. In addition, larger cohorts and further functional studies are needed to reveal the role of autophagy and ATG genes in the pathogenesis of asthma.
We provide evidence showing the association of ATG5 and ATG7 gene polymorphisms with several markers of neutrophilic asthma phenotype, and the different functional effects of ATG5 promoter SNPs on transcriptional activities. The findings suggest a role of ATG genetic polymorphisms and autophagy machinery in neutrophil survival and function, which may contribute to the neutrophilic airway inflammation in asthma.
KEY MESSAGE
1. ATG5 and ATG7 genetic polymorphisms were associated with several makers for neutrophilic asthma phenotype such as sputum neutrophil counts and serum interleukin 8 levels.
2. ATG gene polymorphisms and autophagy machinery could contribute to neutrophilic airway inflammation in asthma pathogenesis.

 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print





