


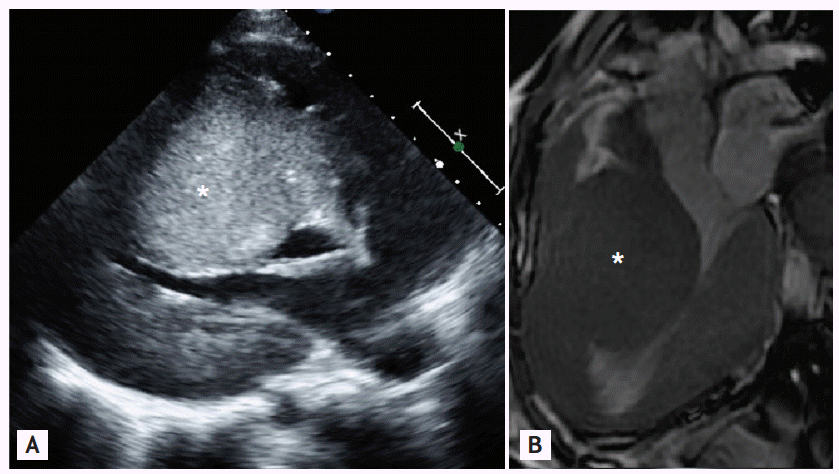
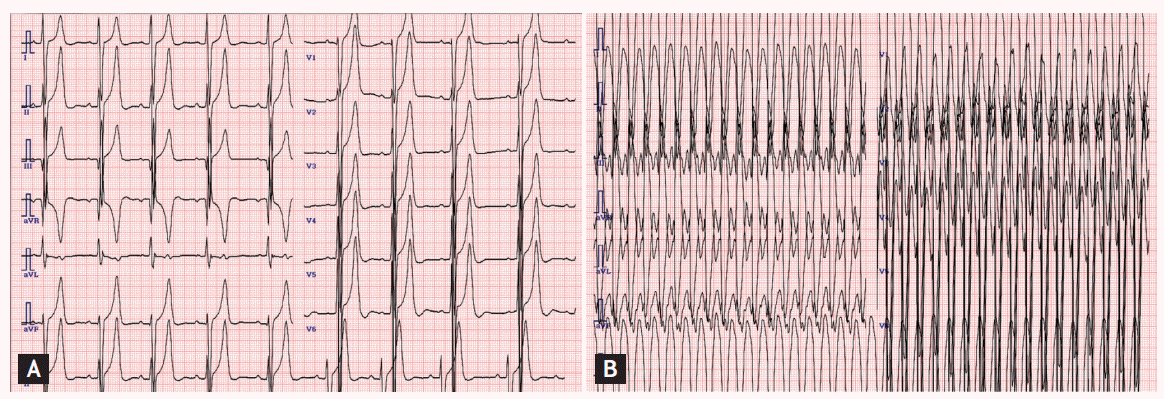
A 14-year-old girl presented to an outpatient clinic for evaluation of dizziness and fainting. She had been diagnosed with congenital hypertrophic cardiomyopathy (HCMP) after birth. Her father was 45 years old and had asymptomatic HCMP (septal wall thickness, 3.0 cm). Her younger sister also had septal hypertrophy (septal wall thickness, 3.5 cm). Her mother had no history of gestational diabetes or corticosteroid exposure. There was no history of sudden cardiac death in their family, including in their siblings. No infiltrative cardiomyopathies such as storage diseases or chromosomal abnormalities were detected. She had been lost to follow-up since the age of 7 years. One year before the herein-described presentation, she visited an outpatient clinic for exertional dyspnea. Echocardiography and cardiac magnetic resonance image (MRI) revealed 6- to 7-cm extreme septal wall hypertrophy with left mid-ventricular outflow obstruction, but without left ventricular outflow tract obstruction (Fig. 1A, parasternal long-axis echocardiography; Fig. 1B, cardiac MRI). Holter monitoring and a treadmill test revealed no abnormal findings. ╬▓-Blockers and calcium channel blockers were administered. At the present admission, an electrocardiogram (ECG) showed sustained ventricular tachycardia that was converted to sinus rhythm after 50-J direct-current cardioversion (Fig. 2A, baseline ECG; Fig. 2B, ECG on second hospitalization). She planned to undergo placement of an implantable cardioverter defibrillator that she had previously refused.
Congenital HCMP is a very rare condition that is caused by a genetic abnormality, maternal diabetes, hyperinsulinism, corticosteroid exposure, and metabolic disorders. A poor prognosis has been reported in symptomatic patients. Although clear evidence has not been provided, septal myomectomy and/or heart transplantation might be considered in refractory cases.
In summary, we have reported a patient with congenital HCMP and extreme septal hypertrophy who survived to adolescence despite symptoms of heart failure without management. We suggest a multidisciplinary approach to this rare condition for careful diagnosis and management.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print





