


To the Editor,
Pulmonary thromboembolism (PTE) ranges from asymptomatic, incidentally discovered emboli to massive emboli causing immediate death. Treatment can reduce the risk of death, and appropriate primary prophylaxis is usually effective. Certain risk factors increase the likelihood for acute deep venous thrombosis and thus PTE. Knowing the risk factors for a given patient should help the physician choose appropriate diagnostic and prophylactic strategies.
Elevated levels of homocysteine (Hcy) are associated with an increased risk for thrombosis [1,2]. We present a case of PTE due to severe hyperhomocysteinemia.
A 66-year-old man was admitted to our hospital because of dyspnea on mild exertion for 3 days. He was a smoker and had a 10-year history of hypertension with medication. He had no known history of diabetes, pulmonary tuberculosis, or hepatitis. He had no medication history except for antihypertensive agents. His family history was non-specific. On arrival, his vital signs included temperature, 36.3Ōäā; blood pressure, 140/90 mmHg; pulse rate, 88 beats/min; and respiratory rate, 32 beats/min.
The patient showed an acutely ill appearance at the time of admission. On physical examination, he had increased jugular venous pressure of 10 cm H2O. Auscultation in the left upper sternal area revealed a normal S1 and a loud P2 with a grade IV systolic ejection murmur in the pulmonic area. No abdominal tenderness or organomegaly was noted. His neurological examination was normal. Peripheral edema was not observed. Arterial blood gas analysis revealed pH, 7.429; pCO2, 33.6 mmHg; pO2, 72.2 mmHg; HCO3-, 21.8 mmol/L; and O2 saturation, 94.5c. His complete blood cell count results were white cells, 11,200/mm3; hemoglobin, 14.5 mg/dL; hematocrit, 40.8%; and platelets, 157,000/mm3. The coagulation profile included activated partial-thromboplastin time, 39.2 seconds (range, 26.5 to 41); prothrombin time, 11.8 seconds (range, 9.8 to 13); fibrinogen, 284.9 mg/dL (range, 180 to 350); fibrinogen degradation products, 8.9 ┬Ąg/mL (range, 0 to 5); and D-dimer, 0.72 mg/L (range, 0 to 0.3). His creatinine level had increased to 1.9 mg/dL (range, 0.5 to 1.2). The lipid profile results showed low density lipoprotein-cholesterol, 121 mg/dL (range, 0 to 120); triglycerides, 133 mg/dL (range, 50 to 150); and lipoprotein(a), 10 mg/dL (range, 0 to 30). The levels of cardiac biomarkers were elevated: troponin I, 0.35 ng/mL (range, 0 to 0.05); C-reactive protein, 1.6 mg/dL (range, 0.1 to 1); and N-terminal prohormone brain natriuretic peptide, 3,106 pg/mL (range, < 262). The estimated creatinine clearance rate using the Cockcroft-Gault formula was 33.92 mL/min/1.73 m2. An electrocardiogram revealed sinus rhythm at a rate of 65 beats per minute and ST-T segment changes over leads III, aVF, and V1-3. Chest radiography revealed no definitive abnormalities such as cardiomegaly, pulmonary congestion, or pneumonic infiltration.
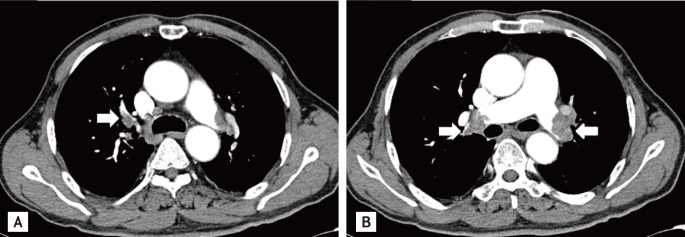
For further evaluation of the dyspnea, we performed transthoracic echocardiography, which revealed a D-shaped left ventricle due to right ventricular pressure overload (Fig. 1A). The right ventricular ejection fraction estimated by tricuspid annular plane systolic excursion had decreased to 33%. Moderate pulmonary hypertension (right ventricular systolic pressure, 61.63 mmHg) was observed (Fig. 1B). These echocardiographic findings suggested acute PTE. We performed chest computed tomography (CT) to confirm the PTE. Multiple PTE in both lobar and main pulmonary arteries were observed on chest CT (Fig. 2). After transferring the patient to the intensive care unit, we performed fibrinolysis with a continuous infusion of tissue plasminogen activator (actylase) 100 mg over 2 hours to dissolve the pulmonary arterial thrombi. An ultrasonogram of the lower extremity venous system revealed no evidence of venous thrombosis. Renal ultrasonography was performed to evaluate the decreased renal function. Multiple cystic lesions in both kidneys were observed on ultrasonography.
We performed further biochemical and immunological examinations to assess the possible thrombophilic cause of the PTE. Vitamin B12 and urine methylmalonic acid levels were within normal limits. The folate level had decreased to 6.31 nmol/L (range, > 12.19). The Hcy level was >50 ┬Ąmol/L (range, 4.72 to 14.05). Protein C, protein S, and antithrombin III levels were within normal limits. The laboratory results for coagulation factors were: factor V, 65% (range, 60 to 140); factor VIII, 169.4% (range, 50 to 150); and factor XII, 37% (range, 60 to 140). The factor V leiden mutation was not detected. Anticardiolipid antibody and antiphospholipid antibody tests were negative. The homozygous point mutation of the methyltetrahydrofolate reductase (MTHFR) gene (677C>T substitution) was identified. Thus, we finally diagnosed PTE caused by severe hyperhomocysteinemia, polycystic kidney disease, and known hypertension.
One-week later, a second transthoracic echocardiography showed remarkable improvement of the impaired right ventricular function and moderate pulmonary hypertension. At 2 weeks after fibrinolysis, a ventilation-perfusion lung scan, performed instead of a follow-up chest CT scan, revealed no significant segmental perfusion defect in either lung field.
The patient received anticoagulation therapy after discharge, with daily supplements of 100 mg of vitamin B6 and 2 mg of folate to prevent recurrence of the PTE. The serum level of Hcy decreased to 12.05 ┬Ąmol/L, and PTE had not recurred after 6 months of clinical follow-up.
A recent revolution has occurred in the field of venous thromboembolism (VTE), which encompasses deep venous thrombosis and PTE. VTE-related deaths have been estimated at 300,000 annually in the United States. Although less common in certain regions such as Asia, VTE is a worldwide problem, particularly in people with known risk factors.
Hcy is a sulfhydryl amino acid derived from the metabolic conversion of methionine, which is dependent on vitamins (folic acid, B12, and B6) as cofactors or cosubstrates. It is clear that hyperhomocysteinemia promotes atherosclerosis and thrombosis in susceptible animal models, but the pathophysiological mechanisms of these effects are less well understood. The overall prevalence of hyperhomocysteinemia in the total population is 5% to 7%. Several clinical studies have reported that elevated levels of Hcy are associated with an increased risk for coronary artery disease and VTE [1,2]. A meta-analysis found that for every increase of 5 ┬Ąmol/L in plasma Hcy concentration, there was an average increase of 60% (odds ratio, 16; 95% confidence interval, 1.1 to 2.34) in the incidence of VTE [2].
Plasma levels of Hcy increase with age, are lower in fertile women than in men, and increase after menopause. Major determinants of plasma Hcy levels include genetic abnormalities, diet (vitamin B2, B6, B12, and folate intake), renal function, cigarette smoking, and coffee and tea consumption.
The most frequent cause of severe hyperhomocysteinemia (characterized by fasting plasma levels of Hcy > 100 ┬Ąmol/L) is a homozygous deficiency of cystathione ╬▓-synthase. The prevalence of this deficiency in the general population is approximately one in 335,000, varying between one in 65,000 (Ireland), and one in 900,000 (Japan). Approximately 5% to 10% of severe hyperhomocysteinemia cases are caused by inherited defects in remethylation. The homozygous point mutation of MTHFR (677C>T substitution), which catalyzes the reduction of methylene-tetrahydrofolate to methyltetrahydrofolate, is the most common inherited defect of the remethylation pathway.
The homozygous MTHFR point mutation leads to a thermolabile enzyme variant with decreased basal enzymatic activity. It has been found in a large proportion of both normal subjects (5%) and patients with vascular disease (17%) in Western countries [3]. Moon et al. [4] reported that the proportion of patients with a homozygous MTHFR C677T gene mutation in Korea was 14% to 20%. Homozygous Korean patients with the MTHFR mutation and coronary artery disease had significantly higher fasting Hcy levels than those in the wild-type enzyme (homozygotes, 18.83 ┬▒ 6.37 ┬Ąmol/L; wild type, 12.36 ┬▒ 3.21 ┬Ąmol/L; p < 0.01) [4]. Three meta-analyses related to the association between MTHFR 677TT (homozygous deficiency) and VTE showed that the TT genotype is associated with increased risk for VTE compared with the wild-type homozygous genotype [2].
It is unclear whether correcting mild to moderate hyperhomocysteinemia reduces the risk for VTE. However, there is clear evidence that the very high VTE risk of patients with severe hyperhomocysteinemia is due to cystathione ╬▓-synthase deficiency; this deficiency decreases dramatically with vitamin supplementation, thereby lowering the very high plasma Hcy levels [5]. No side effects have been reported in trials in which folic acid with or without other vitamins were used for various indications.
In our patient, severe hyperhomocysteinemia was the most possible causative factor for the PTE. The severe hyperhomocysteinemia was associated with old age, smoking, folate deficiency, renal insufficiency, and a homozygous MTHFR C677T gene mutation. With the administration of folate and vitamin B6, the Hcy level decreased to 12.05 ┬Ąmol/L within 3 months, and the patient had no evidence of PTE recurrence during a 6-month clinical follow-up.
This patient with PTE due to severe hyperhomocysteinemia associated with the MTHFR gene mutation was successfully managed with antithrombotics, folate, and vitamin B6. Although it is uncertain whether treatment with vitamins in patients with mild to moderate hyperhomocysteinemia decreases the risk for PTE, we should bear in mind that a patient with a PTE due to severe hyperhomocysteinemia precipitated by a homozygous MTHFR C677T gene mutation can be successfully managed with inexpensive and safe vitamin supplementation.

 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print





