


INTRODUCTION
Benign and malignant teratomas are among the rare tumors of the central nervous system, and constitute only 0.55–0.9% of all intracranial tumors. Intracranial teratomas have a predilection for sites along the midline, namely, the pineal region, the suprasellar region, the celebellar vermis, and the 3rd and lateral ventricles. The pituitary gland has been recognized as an extremely rare site from which teratomas or mixed germ cell tumors can arise, and only one case of intrasellar mixed germ cell tumor originating from the pituitary gland has been reported in the literature. We present a case of a mixed germ cell tumor with a suprasellar and parasellar extension arising from the pituitary gland which underwent transformation from one of benign nature to one of malignant nature.
CASE
A 26-year old primigravida woman was admitted to Severance Hospital for the evaluation of a secondary amenorrhea with galactorrhea. She had been well until 3 years earlier when amenorrhea first developed. Menarche was at the age of 15 with a regular interval of approximately 35–40 days between menstrual period, and a flow of 7 days duration, but menstruation stopped abruptly at the age of 23. Eight months before the amenorrhea was noticed, she had an artificial abortion of her first baby at 4 months IUP. For one year after the amenorrhea was first observed, she had taken shots of progesterone at local clinics and experienced withdrawal bleeding, without effecting a return of the normal menstrual cycle. In the meantime, she had suffered from intermittent headaches and galactorrhea for 3 years. Sudden impairment of visual acuity and diplopia developed 15 days before admission. One month prior to this admission, she had had an appendectomy. One week before this admission, she suddenly became lethargic. With a steroid treatment, she became alert.
Her temperature was 37.2 C; pulse, 76/min.; respiratory rate, 16/min.; and blood presssure 110/70 mmHg. She was 155cm tall and weighed 42 kg. On examination, the patient appeared to be pale, slender, and chronically ill. Her pubic hairs were scanty and axillary hairs were absent. Also, there was an impairment of upward gaze in the left eye.
The urine was normal. The hemoglobin was 10.5 g/dl; hematocrit, 32%; white cell count, 7500/mm3 with 82.4% neutrophils, 14% lymphocytes, and 3% monocytes; reticulocytes count, 0.6%. In the serum, the T3 level was 49.66 ng/dl (normal: 80–220 ng/dl); the T4 level, 2.2 ug/dl (normal: 5–13 ug/dl); free T4, 0.38 ng/dl (normal: 0.68–1.8 ng/dl); estradiol, less than 5 pg/dl (normal: 30–100 pg/dl); prolactin, 6.60 ng/ml (normal: 0–25 ng/ml); HCG, 10.06 mIU/ml (normal: less than 10 mIU/ml); and aFP, 1600 ng/ml. In the 24 hour urine collection, the 17-KS was 5.96 mg (normal: 4–10 mg/day) and the 17-OHCS was 1.86 mg (normal: 3–8 mg/day). Beta-endorphin was undetectable in the serum and in the CSF fluid. A combined pituitary function test was done (Table 1). Ventricular-evoked potential represented a bilateral optic nerve dysfunction.
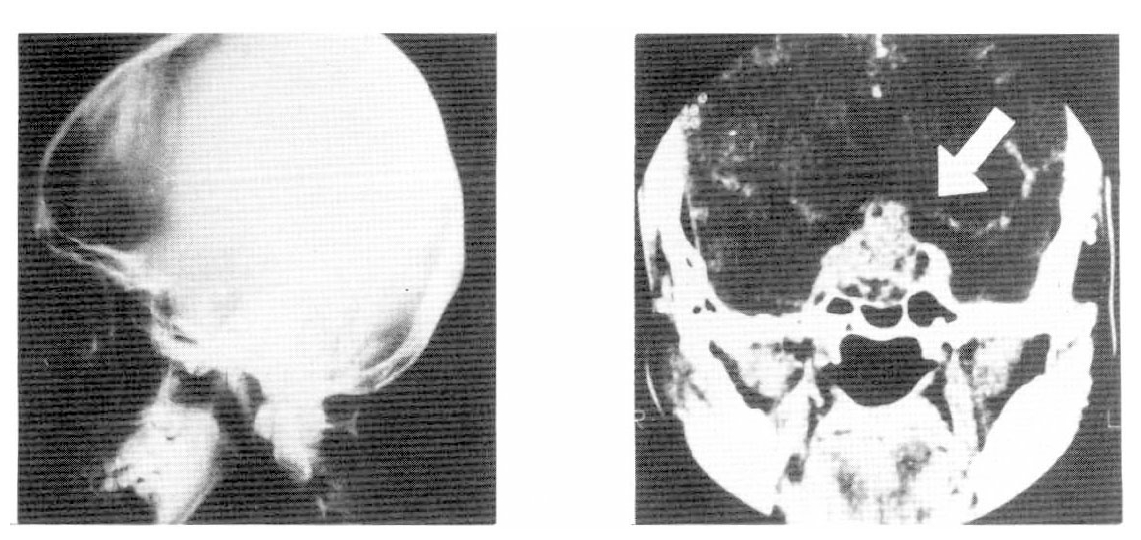
Plain skull X-rays revealed a marked bony erosion of the dorsum of the sella. Cerebral arteriography showed a large pituitary tumor with some parasellar extension and a marked suprasellar extension. A C-T scan showed a pituitary tumor mass with a parasellar and a suprasellar extension, which was greater on the left side. Multiple low density areas were scattered in the tumor mass, representing probable tumor necrosis and hemorrhage (Fig. 1).
1. Surgical Intervention
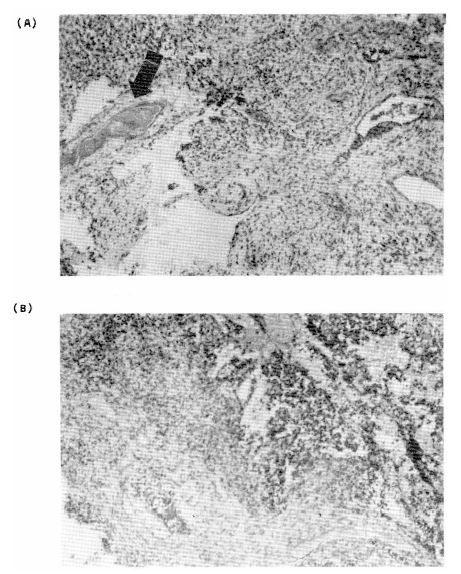
The pituitary fossa was explored through a trans-sphenoidal approaceh. A hard surfaced, encapsulated, somewhat whitish intrasellar mass was removed. Microscopic examination showed neuroectodermal tissue (80%), squamous epithelium, cartilage and skeletal muscle. The neuroectodermal tissue was composed of neuroepithelial elements and mature or anaplastic giant cells (Fig. 2). The post-operative course was complicated by transient diabetes insipidus, and regressive behavior. The follow-up ventricular-evoked potential which was done on the second post-operative day showed improved visual function.
2. First Course of Chemotherapy
On the 17th post-operative day, a course of chemotherapy was started with a PVB regimen under the direction of an oncologist (PVB : cisplatinum 120 mg for 1 days, VP-16 170 mg for 3 days, and bleomycin 30 mg for 1 days). After the chemotherapy, the patient was discharged.
3. Second Course of Chemotherapy
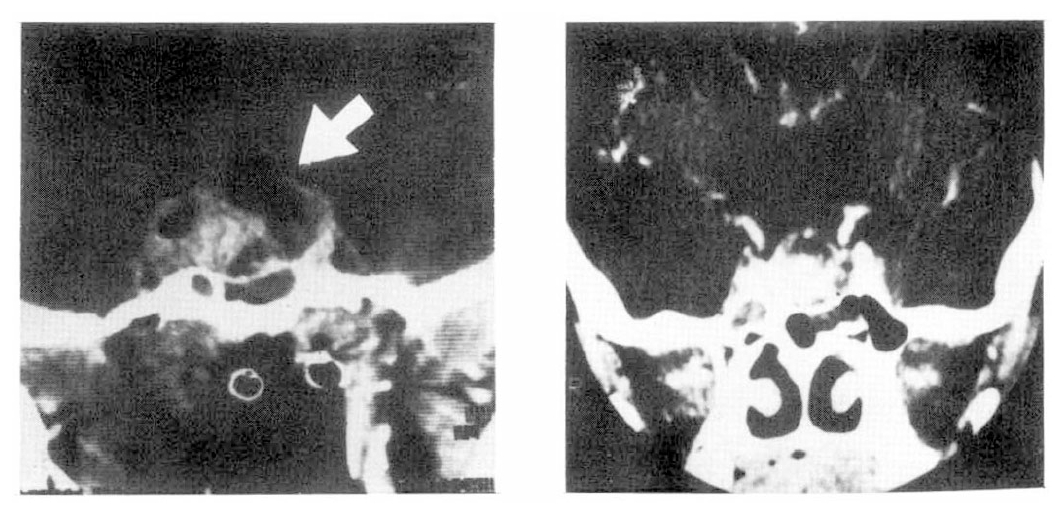
Sixteen days after her first program of chemotherapy, she was readmitted for the second course of chemotherapy. Bilateral hemianopsia had disappeared with an improvement of visual acuity of 20/50 (O.U.). Galactorrhea disappeared, also, but the amenorrhea persisted. Alphafetoprotein became undetectable in the serum and CSF, and there was no elevation on HCG in either. On a follow-up C-T scan, the tumor mass was found to have shrunk (Fig. 3).
4. Radiotherapy
For 5 weeks, after two weeks of rest following the second course of chemotherapy, a course of radiotherapy, with 5,000 rads, was given.
Since the completion of the course of radiotherapy, she has been visiting the hospital periodically for hormone replacement therapy and maintenance chemotherapy with a PVB regimen.
COMMENTN
Teratomas are very rare tumors of the central nervous system constituting only 0.05–0.9% of all intracranial tumors.1) The incidence is higher in children or young adolescents, constituting 2.0% of all intracranial tumors. It seems to be much higher in Japan than elsewhere, representing 2.7–6.7% of all intracranial tumors.2) Most intracranial teratomas have a predilection for the midline, a location with a great potential for the development of tumors from embryonal tissue. The pineal region, suprasellar region, the cerebellar vermis, the third and lateral ventricles are the sites where they originate, in that order.3)
Historically, the classification of midline brain germ cell tumors has been a source of much confusion and disagreement. After Russel postulated that the so-called pinealoma was, in fact, an atypical teratoma or, more correctly germinoma4) a number of reports have been published on cases of germ cell neoplasms in the brain. Currently, brain germ cell tumors are classified as germinma, teratoma, and mixed type. Germinoma, previously identified as ectopic pinealoma, or atypical teratoma, is the most common intracranial germ cell tumor. It is classified as a malignant tumor which consists of cells similar to the primordial germ cells. Teratomas of varying degrees of differentiation are designated as tumors containing the derivatives of all three germ layers, and teratoid tumors as tumors containing only ectodermal and mesodermal components. Mixed types include embryonal carcinoma, choriocarcinoma, and yolk sac tumors. According to Kageyama and Belsky,5) the midline tumors are divided into 3 types according to their originating sites: (1) tumors originating in the pineal region and growing within the optic pathways and the pituitary stalk; (2) tumors originating in the anterior part of the third ventricle and spreading to the chiasm, supra, and intrasellar structure; and (3) tumors originating primarily in the chiasmal region.
Specific types of germ cell tumors produce biological tumor markers, such as alphafetoprotein (aFP) and/or human chorionic gonadotropin (HCG) : choriocarcinoma produces HCG ; endodermal sinus tumor, aFP ; embryonal carcinoma, aFP and HCG. Dysgerminoma and teratoma produce no markers. These tumor markers are useful for classifying the group of tumors, evaluating the response to initial therapy, and also detecting the early recurrence of intracranial tumors.6,7)
When intracranial germ cell tumors occur primarily in the suprasellar region, they usually present a classic triad of hypopituitarism, diabetes insipidus, and visual disturbances.8) The pituitary gland has not been recognized as a site from which germ cell tumor can arise. Mixed germ cell tumors involving the intrasellar region, originating from the suprasellar region, have been reported in 8 cases. But only one case of intrasellar mixed germ cell tumor, arising from the pituitary gland, has been reported in the literature.3) In our case, the patient denied any transient episodes of diabetes insipidus, and that is one of the important reasons why we believe the tumor originated in the pituitary gland and extended rostrally into the brain. The clinical presentation indicated that our patient’s ACTH secretory capacity was not markedly impaired until 1 month prior to admission, since she pulled through the surgical stresses of the appendectomy quite well. The fact that she had experienced sudden bouts of headache and diplopia 15 days prior to admission suggested that a sudden, partial necrosis of the pituitary tumor had occurred, which, of course, had been demonstrated by low density areas found within the tumor mass on the coronal C-T scan. Immediately following admission, she became lethargic, a development which could have been the manifestation of severe cortisol insufficiency after tumor necrosis. Her condition improved on steroid replacement. Diplopia indicated a possible tumor mass involving the cavernous sinus. Furthermore, one of the unusual features of this particular case was the presentation of secondary amenorrhea, galactorrhea, and headache for three years in the absence of other manifestations of critical pituitary insufficiency and visual field defect, which highly suggested that the tumor growth was benign in nature. However, the biopsy specimen of the tumor mass revealed a malignancy with no reference to the endodermal sinus tumor component or the embryonal carcinoma component. Therefore, we concluded that the mass was a malignant tumor transformed from a benign one.
In hormonal studies with a combined stimulation test, performed immediately after admission, only TSH showed a prompt response. LH, FSH, ACTH, and GH exhibited unresponsiveness to the stimulation. On the other hand, the basal level of prolactin showed a mild elevation with a subnormal response to the stimulation. At the same time, in the subsequent serial studies, the prolactin level was not elevated. We concluded that these findings were the results of a PIF interruption rather than a prolactinoma.
In the tumor marker study, the serum aFP level was 1600 ng/ml and the serum HCG level was within the upper normal range. Therefore, even if a careful multisectional histological examination of the entire specimen showed only malignant teratoma elements, there was a possibility that there was an embryonal carcinoma or an endodermal sinus tumor element in the remaining tumor mass. Considering these points, we decided this simple malignant tumor mass should be a mixed germ cell tumor rather than a simple teratoma.
Based on the following radiological findings, with a special consideration that there was no clinical symptom of diabetes insipidus, we believed the primary site of this tumor mass might be the pituitary gland.: (1) in the C-T scan and angiography, severe destruction of the posterior clinoid process with intact anterior clinoid process and a parasellar extension of the tumor mass were found ; (2) the acute angle between the intrasellar mass and the suprasellar mass represented a pressure effect of an intrasellar bulging mass ; (3) with a C-T scan, we could find a bulging diaphragm sellae without destruction ; and (4) in the operative field, the diaphragm sellae was intact and there was no tumor infiltration through the diaphragm.
Combination therapy has become known to be effective in handling malignant teratoma, because formerly, most of the patients with intracranial teratoma died before they reached the age of 25 years.9,10) Therefore, we tried surgical intervention first, and removed most of the anterior portion of the pituitary mass, but the posterior portion and the parasellar portion of the mass was not removed. With regard to a benign teratoma, it is still controversial whether radiation is essential when the tumor mass is completely removed.11) However, in the case of malignant teratoma, it is generally saccepted that radiation treatment should be given to a patient, even if the tumor is totally removed. Since in the case of our patient, we failed to remove all the tumor mass, we gave further management with a combination of chemotherapy and radiotherapy. More improvement in the visual field defect and a shrinkage of the remnant tumor mass were observed after each cycle of chemotherapy, and the level of tumor markers in the serum and the CSF fell to a normal range. After radiotherapy, the patient was put on prednisolone and dessicated thyroid hormone. Our patient has been followed for 6 months, during which time there has been no evidence of tumor extension.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print





