


INTRODUCTION
Recently, the potential role of blood platelets is emphasized on the pathogenesis of coronary atherosclerotic and occlusive vascular disease.
Clinical and experimental evidences indicate that activation and release of platelets may play an important role in the pathogenesis of coronary atherosclerosis and ischemic heart disease. Platelet activation with subsequent formation of platelet aggregates or the generation of thromboxane A2. has been causally implicated in myocardial ischemia.3,4)
PF4 and β-TG, two specific proteins are secreted from the α-granules of platelet during the release reaction. Measurement of plasma levels of these secreted platelet proteins has been suggested as a means for detecting increased platelet activation in vivo.5,8)
We measured the levels of PF4 and β-TG in the peripheral venous blood of patients with acute myocardial infarction or angina pectoris in order to elucidate the relationship between ischemic heart disease and enhanced platelet reactivity in vivo.
SUBJECT AND METHOD
1. Subjects
Patients admitted to the Department of Internal Medicine at Kyung Hee University Hospital from June, 1982 to October, 1983 were evaluated. The study group included 30 patients with acute myocardial infarction and 15 patients with angina pectoris.
2. Method
Blood was drawn through a 20 gauge needle from a vein in the antecubital fossa without tourniquett to avoid the artefactual elevation of PF4 and β-TG levels.
The first 1–2 ml of blood were discarded and the samples were drawn into the tubes each containing EDTA or theophylline and PGE1. The specimens were centrifuged and the top of 0.5 ml of the platelet poor plasma was removed from each specimen, transferred to a clean plastic tube and frozen at −20°C. The PF4 and β-TG levels were measured by radioimmunoassay.
RESULT
There were no significant differences in age, sex, lipid profiles and platelet count between normal controls and patient groups (Table I).
The PF4 and β-TG levels in the patients with acute myocardial infarction were 55.8 ± 12.6 (mean ± S.D.). 128.2 ± 46.1 ng/ml respectively, and those in normal controls were 20.2 ± 15, 67.1 ± 15.8 ng/ml respectively.
In patients with acute myocardial infarction, the PF4 and β-TG levels were significantly higher than those in normal controls (P<0.005). However, in patients with angina pectoris, the PF4 and β-TG levels were not significantly different from those in normal controls (Table 2). The patients with acute myocardial infarction did not differ significantly with normal controls in regard to the distribution of age, sex and coronary risk factors including smoking, hypertension, diabetes and lipid profiles (Table 3).
DISCUSSION
Recent studies have highlighted the importance of coronary atherosclerosis and arterial thrombosis in ischemic heart disease. Occlusive arterial thrombosis results from a focus of platelet aggregation, and altered platelet behavior have been well documented in the patients with ischemic heart disease.1,4)
Until recently, relatively few methods have been available for evaluating platelet function in vivo. These methods can be subject to severe limitation in precision and performance.
In principle it should be possible to measure the platelet release reaction by monitoring the release of a granule marker which will not normally be present in the plasma.
Platelet-specific proteins, PF4 and β-TG which have heparin-neutralizing activity and were secreted during the release reaction, appears to provide a method for investigation of platelet function in vivo.12,16) De Wood et al.17) reported the prevalence of total coronary occlusion in the hours after transmural myocardial infarction. Total coronary occlusion was observed in 110 of 126 patients (87%) who performed coronary arteriography within 4 hours of onset of symptoms. Also, they suggested that coronary thrombi might be the major contributing factor in the pathogenesis of acute myocardial infarction by achieving the limitation of the extent of infarction using the method such as thrombolysis or surgical revasculization.18,19)
Kutti et al.30) observed decreased survival and increased production of platelet in patients with acute myocardial infarction. Handin et al.21) reported that the levels of PF4 in patients with acute myocardial infarction were elevated than those of the normal controls and decreased to the normal range at one week after admission to the hospital.
O’Brien et al.22) have recently reported that patients with acute myocardial infarction have increased β-TG levels and decreased heparin-neutralizing activity. Also Denham et al.23) reported that β-TG levels were elevated in the patients with acute myocardial infarction and the levels were higher in the patients accompanied by mural thrombi.
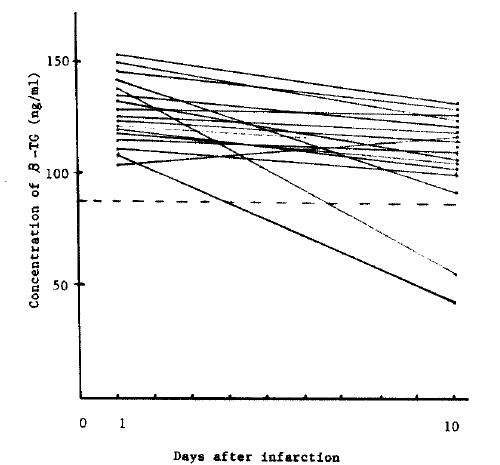
Our results were also in agreement with above data. In our study, we observed that the PF4 and β-TG levels in the patients with acute myocardial infarction were significantly higher compared to those of the normal controls and a pattern of decline to the normal range was shown by day 10 (Fig. 1) (Table 2).
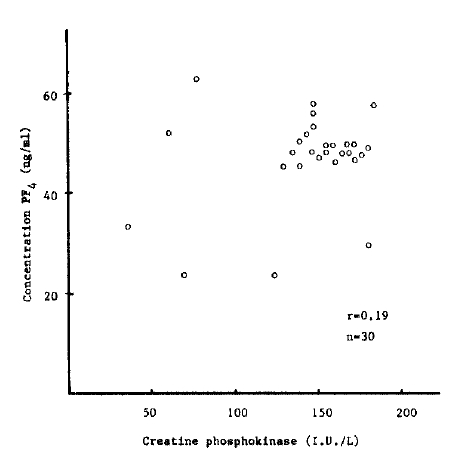
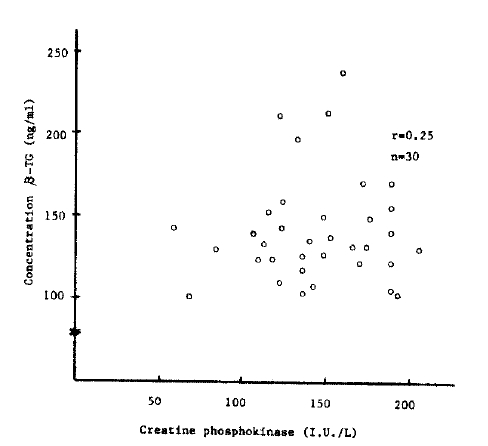
The rate of decline in PF4 levels was steeper than in β-TG levels during admission (Fig. 2); presumably, this might be due to the longer half-life of β-thromboglobulin.24)
These facts provide strong evidence that platelet reactivity increases during the early stage of acute myocardial infarction and the assay of PF4 and β-TG is helpful in studying the state of platelet reactivity in patients with ischemic heart disease.
The assay of such platelet-specific products that are secreted during the platelet-release reaction can obviate many of the uncertainties inherent in studies of platelet function that rely solely on in vitro platelet test or platelet survival techniques.
However, further work will be necessary because not all patients with acute myocardial infarction have the elevated levels of PF4 and β-TG. Boer et al.25) reported that β-TG and thromboxane B2 levels were increased in a minority of the patient with acute myocardial infarction and angina pectoris. Smitherman26) also reported that the β-TG levels were increased in 12 (80%) of 15 patients with acute myocardial infarction.
Hughes et al.27) reported that the β-TG levels were increased in the patients with acute myocardial infarction and angina pectoris than normal controls, but the levels of β-TG did not have significant change during the 10 days after onset of myocardial infarction.
In addition, in the follow-up study significant change of β-TG levels was not shown in spite of recurred infarction or sudden death. Pumphery et al.28) also observed that the β-TG levels in the patients with acute myocardial infarction remained elevated during the first 10 days after disease onset.
These above results may provide that the β-TG levels in the patients with acute myocardial infarction do not reflect the acute stage of thromboembolic state but platelet activation by established atherosclerotic vascular disease.
The PF4 and β-TG levels in the patients with angina pectoris are very variable. Handin et al.21) reported that the PF4 and β-TG levels were not different significantly as compared with normal controls. However, Sobel et al.29) reported PF4 and β-TG levels were increased in patients with unstable angina and higher values were observed when they had symptoms. Smitherman26) observed increased β-TG levels in 4 (16%) of 25 patients with stable angina pectoris and in 23 (59%) of 39 patients with unstable angina pectoris.
In our results, the PF4 and β-TG levels in the patients with angina pectoris were not different significantly from those of the normal controls, so these are in agreement with the result by Handin et al.21)
One cause of this considerable fluctuations and variety in PF4 and β-TG levels may be artefactual elevation of platelet reactivity during the blood sampling and laboratory processing.
So to avoid this artefactual in vitro release Kaplan30) insisted the simultaneous measurement of both PF4 and β-TG. These proteins are present in platelets in similar amounts and are released in similar quantities, but the plasma levels of β-TG exceed the plasma levels of PF4.
This difference in plasma level is presumably due to more rapid removal of platelet factor 4, and there is suggestive evidence that the rapid removal of released PF4 is due to its binding to endothelial cells. It appears that when there is increased release of β-TG and PF4 in vivo, there is an increase in the ratio of plasma β-TG to PF4 compared to that found in normal individuals, whereas in vitro release is responsible for elevated levels, the ratio decreases. Thus, measurements of both proteins in each blood sample will allow distinction between in vivo release and artefactual in vitro release.
Another cause of the considerable fluctuation and variety in PF4 and β-TG levels is the diverse pathogenesis of ischemic heart disease. Acute myocardial infarction is not always accompanied by coronary thrombi, and the pathogenesis of angina pectoris can not be explained only by decreased coronary blood flow evoked from the coronary atherosclerosis.29,31) Platelet thrombosis is probably not involved in the pathogenesis of unstable angina. Coronary spasm may play a role in the development of angina pectoris and myocardial infarction.32)
In addition to the artefactual elevation of PF4 and β-TG levels and diverse pathogenesis of ischemic heart disease, another possible cause is that systemic venous sampling may not be sensitive enough to detect PF4 and β-TG released from platelet in the coronary circulation.28)
Pumphery et al.28,33) reported that no difference was found between patients with no risk factors and those with only one risk factor in the ischemic heart disease, however, patients with two or more risk factors had a significantly elevated plasma β-TG levels. In our study, the patients with acute myocardial infarction did not differ significantly with the normal controls in regard to the distribution of age, sex and coronary risk factors including smoking, hypertension, diabetes and lipid profiles (Table 3).
In conclusion, the measurement of PF4 and β-TG is a useful method to detect the platelet activation in vivo in patients with ischemic heart disease.
Further study will be necessary to determine if continued or recurrent increases in platelet reactivity as manifested by elevated PF4 and β-TG levels are a prognostic marker for an increased risk of morbidity and mortality.

 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print





