


INTRODUCTION
Lymphocytic hypophysitis is a rare inflammatory disorder of the pituitary gland confined to the adenohypophysis. Recently, it has been reported that infundibulohypophysitis is the underlying causality of subsets of central diabetes insipidus. This suggests that lymphocytic hypophysitis is a syndrome of the disorders of the anterior pituitary and the posterior pituitary. Most lymphocytic hypophysitis patients are women. Their symptoms are pituitary mass lesions and/or various degrees of pituitary dysfunction1ŌĆō3). The definite diagnosis of lymphocytic hypophysitis is based on the pathological findings: lymphocytic infiltration in the pituitary gland without granuloma formation4). However, some studies have suggested that the histopathological examination may not be required always to diagnose the hypophysitis if the characteristic clinical features were detected. Most cases have been managed surgically, but steroid therapy is recommended5,6). In this report, we describe a female patient with lymphocytic hypophysitis presenting with diabetes insipidus. She was managed medically.
A CASE REPORT
A 50-year-old premenopausal woman with two children presented with a vague headache and persistent polyuria. She was first seen by a local physician who noted polyuric syndrome and pituitary macroadenoma. She was referred to the Endocrinology Clinic at Chungnam National University. Her bilateral headache was vague, intermittent and had a self-limiting course. She also described a 20-day history of polyuria and polydipsia (8 liters/day). She denied any visual disturbance, galactorrhea, or motor deficits. She did not have the significant medical history and the history of peripartum or postpartum hemorrhage. There was no family history of endocrine or autoimmune disease. Upon admission, the body mass index was 29.5 kg/m2. The results of the physical examination were normal except that the patient was described as being obese. The visual field test evaluated by computerized perimetry was normal.
A laboratory test showed normal blood cell counts. The plasma glucose and serum electrolyte levels was normal. The urine specific gravity was 1.005. Pyuria or hematuria was not detected. The plasma osmolality was 293 mmol/kg (normal: 275ŌĆō295 mmol/kg); urinary osmolality was 73 mmol/kg (normal: 50-1, 400 mmol/kg). During the water deprivation test, the plasma osmolality remained between 289 and 301 mmol/kg and the urine osmolality was between 58 and 224 mmol/kg. After 5 IU vasopressin injection, the plasma osmolality did not alter significantly but the urinary osmolality rose to 534 mmol/kg. The diagnosis of central diabetes insipidus was made and desmopressin in 0.2 mg bid was prescribed.
The baseline hormone test showed the mild elevation of the prolactin level, 60.8 ng/mL. The normal range is 1.5ŌĆō23.5 ng/mL. The thyroid function was normal: free T4, 1.1 ng/mL (normal: 0.7ŌĆō1.9 ng/mL); T3, 1.6 ng/mL (normal: 0.7ŌĆō1.9 ng/mL); TSH, 2.17 U/mL (normal: 0.25ŌĆō4.0 U/mL). The antimicrosomal antibody titer and the antithyroglobulin antibody titer was normal. The basal Cortisol level was 12.4 ╬╝g/dL (normal: 5ŌĆō25 ug/dL), the FSH was 6.6 IU/L (normal: 2ŌĆō10 IU/L), the LH level was 2.8 IU/L (normal: 1ŌĆō8 IU/L), the ACTH level was 19.9 pg/mL (normal: 9.0ŌĆō52 pg/mL), the GH level was 0.14 ng/mL (normal: 0.1ŌĆō7.02 ng/mL), and the Insulin-like Growth Factor-1 level was 224.6 ng/mL (normal: 62ŌĆō350 ng/mL). The combined anterior pituitary function test was performed with insulin 0.3 U/kg, TRH 400 ╬╝g, and LHRH 100 ╬╝g. The result showed partially retained prolactin respone but TSH, FSH, LH, GH and cortisol responses were normal (Table 1).
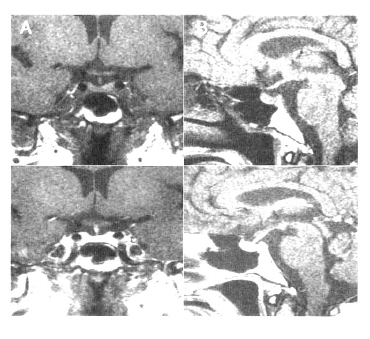
Magnetic resonance imaging of the pituitary gland showed the diffused enlarged pituitary gland with an internal nodule, 1.3 cm in size (diameter). In addition the neurohypophysis high signal on the T1 weighted image was not observed but stalk compression was not detected (Figure 1).
Although her anti-pituitary antibody titer was negative, she was diagnosed as lymphocytic hypophysitis and 1 g methylprednisolone/day was prescribed. After the first 3 days of treatment, she was discharged on a reducing dose of prednisolone. 1 month later, after intravenous administration of methylprednisolone, the second MRI was performed. Her anterion pituitary gland was normal:the size was normal and the nodule disappeared completely. The loss of neurohypophysis signal was persistent, however (Figure 2). At this time of day, her headache disappeared and the polyuric symptoms had improved.
DISCUSSION
Lymphocytic hyphophysitis is a rare inflammatory disorder in the anterior pituitary gland. The disease was first described in 1962, and more than 120 cases have since been reported7). The increased incidence of lymphocytic hypophysitis may be due to the increased awareness6). Although the etiology remains unclear, numerous evidences suggest autoimmune pathogenesis8ŌĆō11). The association with other autoimmune diseases in 30% of the patients such as thyroiditis, hypoadrenalism, parathyroid failure, atrophic gastritis, systemic lupus erythematosus and SjogrenŌĆÖs syndrome may be a diagnostic hint for lymphocytic hypophysitis8ŌĆō11). Lymphocytic hyphophysitis occurs predominantly during pregnancy or in postpartum women. Its occurrence after menopause as well as in men has been reported, however. Aproximately 15% of reported lymphocytic hypophysitis cases are men12ŌĆō14).
Cranial or central diabetes insipidus may be idiopathic. Alternately, the disease may be inherited as an autosomal dominant trait or an X-linked recessive trait15). In most cases, cranial diabetes insipidus is secondary to the disturbance of the hypothalamic-neurohypophyseal system, which may be due to a wide range of conditions such as trauma, infection, tumors, and granulomatous disease16). Although lymphocytic hypophysitis may rarely present with diabetes insipidus, the sudden onset of diabetes insipidus is a hallmark of lymphocytic hypophysitis6). Lymphocytic infundibulohypophysitis has been suggested to be the underlying cause in some cases previously labeled ŌĆ£idiopathicŌĆØ diabetes insipidus cases15). In lymphocytic infundibulohypophysitis, MR imaging may demonstrate the thickened pituitary stalk or the enlarged neurohypophysis16). In addition, the loss of the hyperintense signal of the normal neurohypophysis on contrast enhanced T1 weighted imaging may be detected. The anterior pituitary is spared, however. This distinguishes lymphocytic infundibulohypophysitis from lymphocytic hypophysitis radiologically17).
The possible explanations for diabetes insipidus with lymphocytic hypophysitis are: (1) the extension of the mass into the hypothalamus resulting in the suppression of vasopressin synthesis and/or (2) the direct involvement of the pituitary and/or neurohypophysis with inflammatory reaction causing the destruction of the hypothalamic pathways18,19).
Other clinical manifestations such as headache, chiosm compression, and diplopia lack specificity6). In addition to the partial or total hypopituitarism, hyperprolactinemia may be present in lymphocytic hyphophysitis patients. Furthermore, the elevated prolactin level may represent an endocrine marker in many lymphocytic hyphophysitis patients1). Stalk compression resulting reduced dopamine delivery to the anterior pituitary may be reason for the hyperprolactinemia. However, an autoimmune mechanism involving the production of stimulating_antibodies by plasma cells may lead to increased prolactin secretion7,20). Patients with presumed SheehanŌĆÖs syndrome but without clear history of postpartum hemorrhage or sepsis may have lymphocytic hypophysitis.1 SheehanŌĆÖs syndrome is cuased by the ischemic pituitary necrosis due to severe postpartum hemorrhage and almost always characterized by the empty sella. Such symptoms rarely present in diabetic insipidus21,22). As the prolactin deficiency has been detected in SheehanŌĆÖs syndrome, the evaluation of the prolactin level has been used as its screening procedure. However, hyperprolactinemia has been reported in one SheehanŌĆÖs syndrome patient23). Our patient lacks the typical medical history suggesting SheehanŌĆÖs syndrome, such as postpartum hemorrhage, agalactorrhea, and amenorrhea during the postpartum period. This clearly rules out SheehanŌĆÖs syndrome in our patient.
In lymphocytic hypophysitis, MRI reveals the symmetric enlargement of the sellar content with the thickened pituitary stalk, that are homogenously enhancing. However, the symmetric enlargement of the sellar content, the signal alteration on the native T1 weighted, the inhomogenous contrast enhancement, the involvement of the carvernous sinus and hypothalamus, the empty sella, or no pathologic findings have been described6,7).
None of the features described above is unique to lymphocytic hypophysitis. Some uncertainty thus will always exist without the pathologic confirmation. Our patient was a premenopausal woman with vague headache, mild hyperprolactinemia and the 20-day onset of diabetes insipidus. No evidence of the stalk compression on the MRI has been detected. However, we have observed the diffused enlargement of the anterior pituitary gland with an internal nodule and the disapperance of the normal bright spot in the posterior pituitary gland. In our patient, the hyperprolactinemia might be due to the autoimmune reaction and the diabetes insipidus may be due to direct involvement of neurohypophysis.
The hypophysitis may be either primary or secondary. In the secondary hypophysitis, etiologic agents or defined systemic diseases have been implicated as the causality. The causative agent of the primary hypophysitis is currently unknown. Three distinct clinicopathological entities, however, have been described: lymphocytic hypophysitis, granulomatous hypophysitis, and xanthomatous hypophysitis24). Currently, it is unclear whether these three conditions are truly distinct entities or merely different manifestations of the same disease. They share clinical and radiological features and the reliable way to distinguish them without proper histological examinations is not available25. Nevertheless, they are important in making a differential diagnosis of the sella mass, and conservative management may lead to their resolution without surgery24).
It has been suggested that if the inflammation of the pituitary gland were left untreated, lymphocytic hypophysitis may worsen because of the progressive pituitary insufficiency. However, it may improve spontaneously19). In view of our recent experience, the presumptive diagnosis of lymphocytic hypophysitis may be established by a trial of high dose methylprednisolone with close monitoring. The follow up at short intervals is required to detect the possible relapse in time. The indications for surgery are the presence of the gross chiasm compression, the ineffectiveness of corticosteroid therapy, and the inconclusive diagnosis of lymphocytic hypophysitis by conservative evaluations6). In our patient, the high dose methylprednisolone pulse therapy induced the complete remission of the pituitary enlargement and improved her subjective symptoms in short time.
In conclusion, lymphocytic hyphophysitis is a rare disorder that should be considered in the diagnosis of pituitary mass. The mass may extend to the suprasellar region. In addition, lymphocytic hyphophysitis should be suspected in patients with the empty sella, hyperprolactinemia, hypopituitarism and cranial diabetes insipidus. Furthermore, we recommend the high dose methylprednisolone rather than surgery: it can resolve the cellar mass and improve the endocrine dysfunction such as the polyuric symptoms.

 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print





