


INTRODUCTION
Ischemic acute kidney injury (AKI) is an important clinical disorder affecting a large number of patients worldwide and is a major risk factor for chronic kidney disease (CKD) and end-stage renal disease [1]. Any clinical conditions that can result in an interruption of renal blood flow—such as intravascular volume depletion, reduced cardiac output, and vasodilatory or vasoconstrictive disorders—can cause ischemic AKI [2,3]. Given its high prevalence and mortality, despite continued and substantial improvement in critical care and dialysis techniques, improving ischemic AKI outcomes is challenging [4,5]. Therapies that reduce injury or enhance adaptive repair are not available [5]. Among various cellular and molecular pathways involved in its pathogenesis, inflammation orchestrated by kidney immune cells affects both injury and repair processes [6]. Normal kidneys of human and animals possess considerable amounts of resident immune cells [7,8]. Indeed, even germ-free (GF) mice that have not been exposed to microbiota have abundant kidney resident immune cells, which are related to ischemic AKI pathogenesis, indicating that they play pathogenic roles under sterile conditions [9]. In ischemic AKI, endogenous molecules released from stressed or injured cells trigger activation and proliferation of kidney immune cells, contributing to the progression of injury. This process resembles immune responses to microbial pathogens and is thus termed sterile inflammation [10,11]. Among the immune cells participating in this process, T cells, a major component of the adaptive immune system, play crucial roles as major effectors and regulators [12]. Although most evidence of their role in AKI is from animal studies [13], continued clinical experience of AKI in patients receiving T cell-targeted immune check point inhibitor or chimeric antigen receptor T cell therapy highlights their pathogenic roles in human AKI [14,15]. A rigorous mechanistic understanding of AKI would facilitate development of novel therapeutic targets. Here we review the mechanisms of ischemic AKI, focusing on the role of T cells during the injury phase and recovery process based on experimental studies, emphasizing recent literature.
ROLE OF T CELLS DURING EARLY RENAL INJURY IN ISCHEMIC AKI
Studies using murine ischemia-reperfusion injury (IRI) models have provided the most mechanistic evidence of T cells in ischemic AKI (Table 1) [7,9,16–42]. In contrast to the classical concept of immunity, which describes adaptive immune cells as relatively late responders in the injured site, T cells traffic into post-ischemic kidneys at a very early time point and coordinate innate immunity [16,17]. An in vitro study of hypoxia-reoxygenation demonstrated T cell adhesion to tubular epithelial cells [18].
During the early phase of IRI, T cells play pathogenic roles by contributing to the injury process, as demonstrated using mice lacking T cells. Both T cell-deficient mice (nu/nu mice) and CD4, CD8 double-knockout (KO) mice were protected from IRI [18,19]. A study using a whole body IRI model, by inducing cardiac arrest followed by resuscitation, reproduced these findings, showing significant protection in T cell-deficient mice with reduced expression of intercellular adhesion molecule 1 (Icam1) [20]. There was substantial functional and structural protection from IRI in T cell receptor (TCR) αβ-deficient mice, with a reduction of tumor necrosis factor α (TNF-α) and interleukin 6 (IL-6) expression in the post-ischemic kidneys [21]. A subsequent study using genetically engineered DO11.10 transgenic mice demonstrated that diverse TCR repertoires are essential for the initiation of T cell activation but, once activated, TCR diversity is likely to have less impact on their pathogenic effect [22].
Importantly, T cell costimulatory molecules, emerging therapeutic targets in oncology, play important roles in the pathogenesis of ischemic AKI. When T cells recognize antigens through TCR, binding of CD28 to B7 (also known as CD80 and CD86 for B7-1 and B7-2, respectively) on antigen-presenting cells functions as a costimulatory pathway, amplifying the TCR signal to activate T cells [43]. A negative costimulatory molecule on T cells, cytotoxic T lymphocyte-associated protein 4 (CTLA-4), outcompetes B7 binding, dampening T cell activation [43]. Blockade of the CD28-B7 costimulatory pathway by CTLA-4-Ig administration in cold IRI reduced renal dysfunction and inhibited T cell infiltration into the post-ischemic kidneys [44]. In line with this finding, adoptive transfer of CD28-deficient T cells into T cell-deficient mice (nu/nu mice) did not reinduce the injury phenotype [19]. Programmed death-1 (PD-1) expressed on T cells is another negative costimulatory molecule, which has two ligands, programmed death ligand 1 (PD-L1) and PD-L2 [43]. Blocking of the PD-1 pathway using an antibody against PD-L1 or PD-L2, or genetic deficiency of either, led to deterioration of structural and functional renal injury in post-ischemic kidneys [23]. Blocking both PD-L1 and PD-L2 led to upregulation of proinflammatory gene expression in post-ischemic kidneys, including Il6, C-X-C motif chemokine ligand 1 (Cxcl1), and Icam1 [23]. Importantly, PD-L1 is expressed on renal tubular epithelial cells as well as on immune cells, and interferon γ (IFN-γ) induces upregulation of PD-L1 on renal tubular epithelial cells [45]. Because bone marrow transfer from wild-type (WT) mice to PD-L1 KO mice failed to induce protection, the PD-L1 expression on renal tubular epithelial cells, rather than immune cells, may serve as a major protective mechanism in ischemic AKI [23].
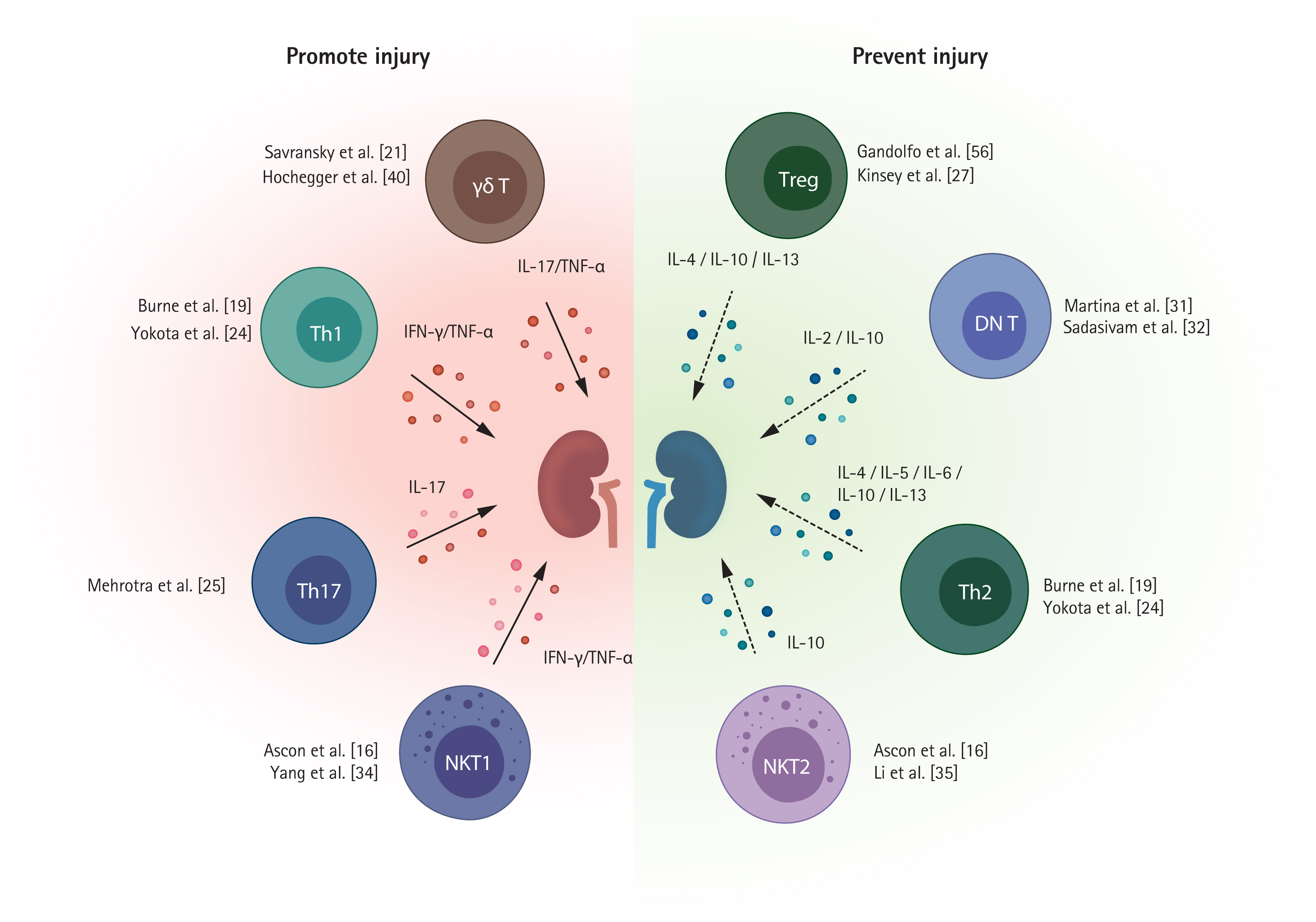
Several T cell subpopulations exhibit distinct roles during the injury process (Fig. 1). Below we discuss the major subpopulations of T cells.
CD4+ T cells
CD4+ T cells are major mediators of early renal injury, supported by reports that mice lacking CD4+ T cells are more protected from IRI than those lacking CD8+ T cells [19]. CD4+ T cell adoptive transfer to CD4+ T cell-deficient mice reconstituted renal injury [19]. Sphingosine-1-phosphate (S1P) and S1P receptor (S1PR) signaling is required for T cell trafficking to peripheral tissues from lymphoid organs, and S1PR1 selective agonists block T cell egress from lymphoid tissue [46]. S1PR1 selective agonist pretreatment before IRI exerted a renoprotective effect by inhibiting CD4+ T cell infiltration into post-ischemic kidneys [17]. Renal CD4+ T cells can undergo polarization into T helper (Th) cells, which comprise three major subsets—Th1, Th2, and Th17.
Th1/Th2 cells
Th1 cells secrete IFN-γ and TNF-α and are regulated by the transcription factor T-bet. Th2 cells produce IL-4, IL-5, IL-10, and IL-13 and are regulated by the transcription factor GATA binding protein 3 (GATA3) [47]. Th1 cells and Th2 cells are likely to have opposite effects on the pathogenesis of ischemic AKI: Th1 cells play a pathogenic, and Th2 cells a protective, role [24,48]. The signal transducer and activator of transcription 4 (STAT4) gene and STAT6 gene are responsible for Th1 differentiation and Th2 differentiation, respectively. Mice lacking STAT4 were protected from IRI, whereas mice lacking STAT6 showed more severe injury after IRI [24]. The dendritic cell sphingosine 1-phosphate 3 (S1P3) pathway is involved in Th1 polarization, and adoptive transfer of S1P3-deficient dendritic cells protected against IRI by inducing Th2 polarization [48].
Th17 cells
Th17 cells are regulated by the transcription factors STAT3 and retinoic acid receptor-related orphan receptor-γt (RORγt) and produce IL-17, which recruits innate immune cells [49,50]. Th17 cell polarization is mainly driven by the STAT3 activating cytokines IL-1 and IL6, and transforming growth factor β (TGFβ) contributes by suppressing Th1 or Th2 polarization [51,52]. Along with their detrimental roles in autoimmune disorders and transplantation, they play pathogenic roles in ischemic AKI [25,53]. IL-1 secreted by renal dendritic cells and macrophages promotes Th17 differentiation and activation following kidney injury [54]. Increased kidney Th17 cells were observed within the first 3 days after IRI and decreased to a comparable steady state within 7 days of renal recovery [25]. More specifically, calcium channel Orai1-expressing Th17 cells are involved in IL-17 production in IRI, contributing to tissue damage [26].
CD8+ T cells
CD8+ T cells can differentiate into cytotoxic effector cells with upregulation of TNF-α and IFN-γ [7], but they have been less studied than CD4+ T cells in AKI. CD8+ T cells may contribute to renal injury by producing INF-γ in renal IRI [7]. However, compared to CD4+ T cells, CD8+ T cells are likely to have a minor effect on renal IRI. Unlike CD4-deficient mice, mice lacking CD8+ T cells showed a comparable level of injury to WT mice after IRI [19]. The roles of CD8+ T cells were evaluated using GF mice. GF status was linked to a reduced proportion of effector memory phenotypes (CD44high CD62Llow) of CD8+ T cells and worsened kidney injury after IRI with enhanced CD8+ T cell trafficking into post-ischemic kidneys. Therefore, naive CD8+ T cells associated with underexposure to microbial stimuli appear to have a pathogenic role in post-ischemic kidneys [9].
Regulatory T cells
Regulatory T cells (Tregs) have CD4 and CD25 as surface markers and upregulation of nuclear transcription factor forkhead box P3 (FoxP3) [55]. A small percentage of CD4+ T cells are Tregs in normal mouse kidneys, accounting for only 3% to 4% of total CD4+ T cells [7,56]. Contrary to the deleterious role of conventional CD4+ T cells, Tregs exert protective effects by suppressing proinflammatory responses despite their small numbers [57].
Partial Treg depletion with an anti-CD25 antibody (PC61) in WT mice before IRI resulted in more severe injury, with increased neutrophil and macrophage trafficking and upregulation of Il6, Tnfα, and Tgfβ in post-ischemic kidneys [27]. Adoptive Treg transfer to mice lacking lymphocytes (Rag1 KO mice) prior to IRI showed a protective effect. An anti-inflammatory cytokine, IL-10 produced by Tregs, appears to be an important mediator for their protective mechanism. IL-10-deficient Treg adoptive transfer did not show a protective effect in Rag1 KO mice [27]. A study using a more complex IRI model to simulate ischemic preconditioning demonstrated that Tregs are involved in the protective mechanism of the ischemic preconditioning [28].
An important mechanism by which Tregs exert an anti-inflammatory effect on AKI is adenosine generation by CD73, to which adenosine 2A receptor (A2AR) on Tregs contributes in an autocrine manner [29]. Notably, PD-1 expression on Tregs is likely to be vital for their anti-inflammatory function. Activation of the A2AR signaling pathway enhanced PD-1 expression on Tregs, and PD-1 depletion on Tregs abolished the protective effect [29]. Blocking PD-1 ligands using an anti-PD-L1 or PD-L2 antibody before Treg adoptive transfer also negated its protective effect on IRI [23].
Because the complex of IL-2 and a particular form of IL-2 antibody (JES6-1) reportedly preferentially expanded Tregs, an IL-2/anti-IL-2 complex (IL-2C) has been tested in renal IRI [30]. IL-2C induced expansion of Tregs in kidneys and ameliorated renal injury. Treg depletion by anti-CD25 antibody abolished the protective effect of IL-2C [30].
The pro-reparative role of Tregs during the recovery phase of ischemic AKI will be reviewed separately in the next section.
Double-negative αβ T cells
Double-negative (DN) T cells (TCRαβ+, CD4−, CD8− T cells) are an important subset of kidney resident αβ T cells [7]. Although rare in lymphoid organs, DN T cells represent approximately 20% to 25% of kidney αβ T cells in normal mouse kidneys and post-ischemic kidneys [7,31]. Considerable proportions of DN T cells were also found in human ischemic kidneys [7,31]. DN T cells were previously thought to originate from CD4+ or CD8+ T cells by losing their CD4 or CD8 receptor [58,59], but are now considered a discrete subset distinguished from conventional CD4+ or CD8+ T cells in kidneys because DN T cells were present in β2m-deficient mice and major histocompatibility complex (MHC) II-deficient mice [32]. IL-2 is required for activation and proliferation of DN T cells [32]. Kidney DN T cells contain PD-1+ subset and NK1.1+ subset in both human and mice [32]. DN T cells have different immunophenotypic profiles with higher expression of CD28, CD44, and CD69 compared to CD4+ and CD8+ T cells in a steady state and significantly higher proliferation capacities [31]. Thus, DN T cells are expected to play distinct roles in AKI pathophysiology.
Mouse anti-thymocyte globulin treatment increased the DN T cell proportion in the post-ischemic kidney [33]. DN T cells increase rapidly within 24 hours after IRI and likely have an IL-10-dependent protective function in the early injury process [31]. An adoptive transfer of DN T cells from Faslgld mice before IRI showed a protective effect, and IL-10 neutralization with an anti-IL-10 receptor antibody abrogated the protective effect [31].
The PD-1+ DN T cell subset was predominant following IRI, whereas the NK1.1+ subset decreased; thus, PD-1+ DN T cells may be important in the pathogenesis of ischemic AKI. Based on the higher expression of PD-1 on DN T cells from human kidney tumor tissue, DN T cells may be relevant targets of immune checkpoint inhibitor therapy [32]. Few studies have addressed DN T cells, so further research is needed.
Unconventional T cells
Unconventional T cells—including γδ T cells and natural killer (NK) T cells—are different types of T cells from conventional CD4+ and CD8+ T cells in their TCR usage. Unconventional T cells present as minor T cell subsets in the kidney [60]. Because unconventional T cells can act in a non-MHC-restricted manner, a feature of innate immunity, they are also referred to as ‘innate-like T cells’ in the literature [60]. These unconventional T cells are much less understood and have extensive interspecies differences unlike conventional T cells, hampering studies using experimental animal models.
Natural killer T cells
NK T cells can directly recognize antigens presented on CD1d molecules from antigen-presenting cells, thus are defined as CD1d-restricted T cells. NK T cells reportedly infiltrate post-ischemic kidneys at a very early time point [16]. NK T cells were also found in renal tissue of patients with acute tubular necrosis, but are rare in normal human kidney specimens [34]. Given the capability of NK T cells to become rapidly activated, resulting in cytokine production (including INF-γ) and recruitment of innate immune cells [35], they are considered a major effector and modulator cell type in the very early phase of IRI. NK T cells are likely to mainly have a deleterious effect on ischemic AKI [35]. NK T cell depletion using an anti-NK1.1 antibody and mice lacking NK T cells were protective against ischemic AKI [35]. Blocking NK T cell activation using an anti-CD1d antibody ameliorated renal injury [35]. Tolerogenic dendritic cells treated with an A2AR agonist exerted a protective effect on IRI by suppressing NK T cell production of IFN-γ [36]. In addition, IL-33 is a mediator of NK T cell recruitment and activation in post-ischemic kidneys [37], whereas the hypoxia-inducible factor (HIF)-2α and A2AR pathways exert protective effects by restricting NK T cell infiltration and activation [38].
There are two subsets of NK T cells, type I NK T cells and type II NK T cells [61]. Because type I NK T cells have an invariant TCR α chain, they are also referred to as ‘invariant NK T cells,’ whereas type II NK T cells are called ‘diverse NK T cells’ based on their more diverse TCR α- and β-chain expression [61].
Type I NK T cells can be stimulated by α-galactosylceramide (α-GalCer), whereas type II NK T cells cannot [61]. α-GalCer injection in normal mice induced AKI by activating type I NK T cells in the kidney [39]. The TNF-α/FAS ligand pathway and perforin-mediated pathway are related to NK T cell-mediated glomerular injury and tubular injury, respectively [39].
Type II NK T cells are likely to have opposite roles to type I NK T cells in ischemic AKI. They can be stimulated by sulfatide and nonlipid antigens. Sulfatide injection into WT mice and type I NK T cell-deficient mice ameliorated renal injury following IRI. Sulfatide injection to CD1d-deficient mice (mice lacking both type I and II NK T cells) did not show a protective effect [34]. This renoprotective function in renal IRI is mediated via the HIF-1α and IL-10 pathways [34].
γδ T cells
γδ T cells exacerbate ischemic AKI—γδ T cell deficiency or depletion has a protective effect on renal IRI [21,40]. γδ T cells were identified in human healthy kidney tissues as a minor fraction of T cells, and their number in the kidney was correlated negatively with the estimated glomerular filtration rate and positively with the degree of fibrosis in patients with CKD [62]. γδ T cells infiltrate damaged kidneys from the blood following ischemic AKI; a rapid drop in circulating γδ T cell levels followed by increased γδ T cells in the kidneys was observed in human and mouse AKI [40,41]. Although the significance of γδ T cells in the pathogenesis of ischemic AKI is unclear, given their minor fraction and less protective effect on γδ T cell deficiency than αβ T cell deficiency, they are likely to have less influence on AKI pathophysiology compared to conventional αβ T cells [40,41].
ROLE OF T CELLS IN RECOVERY OR AKI TO CKD TRANSITION
Given the lack of established real-time biomarkers for AKI, clinical interventions mostly become available after the establishment of renal injury as well as in the recovery phase [63]. Moreover, therapeutic approaches that inhibit inflammatory processes during the injury phase may disrupt the adaptive repair process [6]. Thus, understanding and targeting the T cell-mediated repair process has high clinical importance, but few studies have focused on the recovery phase. Here, we review studies that reported long-term renal outcomes or conducted interventions after renal injury (Table 2) [28,30,56,64–70].
CD4+ and CD8+ T cells
Chronic T cell infiltration into post-ischemic kidneys was investigated using IRI models. The study based on immunohistochemistry findings demonstrated increased CD4+ T cells at 6 weeks after IRI, and there was enhanced expression of IL-1 and C-C motif chemokine ligand 5 (CCL5) [64]. Subsequent studies based on flow cytometry with isolated kidney infiltrating mononuclear cells showed consistent findings [65]. Activated phenotype (CD69+) and effector-memory phenotypes (CD44high CD62L−) of T cells were the dominant phenotypes during the long-term recovery phase [65]. Moreover, expression of inflammatory cytokines and chemokines was enhanced in post-ischemic kidneys until 6 weeks [65]. Thus, T cell activation and an inflammatory microenvironment following ischemic AKI are likely to be maintained chronically and play important roles in tubular regeneration or the AKI to CKD transition. A study using single-cell RNA-sequencing (scRNA-seq) showed a marked increase in the T cell compartment at the transcriptomic level during the recovery phase. Among T cell subtypes, Th17 cells were gradually increased after IRI until 4 weeks [66].
Transcriptomic analyses of sorted T cells from post-ischemic kidney tissue showed that their Ccl5 gene expression was consistently upregulated until 6 weeks after IRI [67]. Also, C-C motif chemokine receptor 5 (CCR5) blockade using an anti-CCR5 antibody improved renal outcomes functionally and structurally following ischemic AKI [67]. This finding was reproduced by scRNA-seq data showing the Ccl5-Ccr5 interaction was significantly associated with failed repair of tubular cells following IRI [71].
Extrarenal T cells are also activated until the recovery phase of ischemic AKI. Enhanced INF-γ secretion was observed from splenic T cells at 6 weeks after IRI [64]. Adoptive transfer of splenic T cells isolated from post-IRI donor mice at 6 weeks after IRI induced albuminuria in naïve recipient mice at 12 weeks after the injection [68]. This finding suggests that the inflammatory property of extrarenal T cells is chronically maintained following renal IRI, which may affect renal recovery.
Regulatory T cells
Tregs not only prevent damage in the early phase but also have a pro-reparative function by preventing fibrosis development during the recovery phase. A late Treg (CD4+ CD25+ FoxP3+) trafficking into post-ischemic kidneys were demonstrated [56], and IL-10 expression on Tregs were unregulated during recovery phase [28]. A scRNA-seq based study reproduced the finding of late Treg accumulation at the transcriptomic level [66]. Notably, it was also revealed that a set of reparative genes in Tregs were expressed at the highest levels at later time points following IRI, indicating their important roles in the repair process [66].
Adoptive transfer of Tregs isolated from spleen into post-IRI mice at 24 hours after IRI mitigated structural renal injury and enhanced tubular regeneration at 10 days after IRI, accompanied by a reduction of the CD4+ T cell TNF-α production [56]. Treg depletion using an anti-CD25 antibody starting at 24 hours after IRI suppressed tubular regeneration and increased T cell TNF-α and IFN-γ production [56]. Treg expansion using an IL-2C reduced renal fibrosis at 4 weeks after IRI. Also, Treg depletion with an anti-CD25 antibody abrogated the beneficial effect of IL-2C [30]. A combination of IL-2C and IL-33 administration markedly increased Tregs in post-ischemic kidneys and reduced renal injury with less fibrosis at 4 weeks after IRI [66].
Although theoretically promising, the use of immunosuppressive agents in AKI has failed to achieve favorable outcomes. However, our understanding of the role of Tregs has increased. Mycophenolate mofetil worsened tubular injury and reduced Treg expansion in post-ischemic kidneys at 10 days after IRI, highlighting the reparative role of Tregs during renal recovery [69]. Because early initiation of mammalian target of rapamycin (mTOR) inhibitors can retard wound healing and delay renal recovery [72,73], delayed administration of rapamycin was evaluated. However, it also increased albuminuria following IRI [74]. Notably, rapamycin induces Treg expansion and IL-10 upregulation in vitro. Thus, rapamycin ex vivo-treated Tregs were adoptively transferred at 24 hours after IRI. There was less tubular injury and lower expression of fibrosis markers in the rapamycin ex vivo-treated Treg transfer group at 14 days after IRI [70].
There was a clear transcriptional difference between Tregs in a regenerative environment and those in a profibrotic environment. Tregs seem to have complicated tissue specific functions, warranting further research [66].
T cell and distant organ crosstalk during ischemic AKI
Although renal failure can be managed with renal replacement therapy, unlike other organ failures, AKI-associated mortality remains high even when prompt dialysis is available [75,76]. Thus, increased mortality in patients with AKI cannot be solely explained by reduced renal clearance per se, and mechanisms beyond that are postulated [77]. One is the inflammatory response by kidney T cells and other immune cells, which has promise as a therapeutic target to reduce AKI-associated mortality. In this section, we briefly review the role of T cell-mediated systemic inflammatory effects on distant organs.
Pulmonary inflammation is a common AKI-related distant organ damage [77]. AKI induces acute lung injury or acute respiratory distress syndrome (ARDS) [78]. Beyond volume overload due to reduced renal clearance, the systemic inflammatory effect of AKI contributes to AKI-associated pulmonary complications [79]. Higher levels of circulating inflammatory cytokines were found in patients with AKI-related ARDS than in ARDS patients without AKI [80]. A landmark experimental study demonstrated that renal IRI generated distinguishable lung transcriptomic profiles (including upregulated proinflammatory and proapoptotic pathways) from those induced by bilateral nephrectomy [81]. T cells traffic into the lungs and cause pulmonary endothelial apoptosis through the caspase 3 pathway during ischemic AKI [82]. T cell-deficient mice did not show caspase-3 activation, and adoptive transfer of T cells from WT mice into T cell-deficient mice reconstituted pulmonary endothelial apoptosis [82]. Circulating IL-6 is a major pathogenic mediator of lung injury following AKI and induces upregulation of pulmonary CXCL1 expression [83].
AKI is also associated with adverse cardiac events, and interactions between heart and kidney diseases are clinically defined as cardiorenal syndrome [84]. Among the five subtypes of cardiorenal syndrome, the pathophysiology of cardiorenal syndrome type 3, acute cardiac dysfunction caused by an abrupt and primary worsening of kidney function, is less studied [78]. T cell-mediated mechanisms including systemic inflammation and activation of neurohormonal pathways may be involved [78,85]. This concept is supported by experimental evidence showing that renal IRI induced cardiac cell apoptosis but bilateral nephrectomy did not [86]. This cardiac injury was accompanied by increased levels of TNF-α and IL-1 in heart tissue and the systemic circulation [86]. Importantly, the activation of neurohormonal pathways such as the renin-angiotensin-aldosterone system (RAAS) in AKI promotes cardiac inflammation and fibrosis [87]. RAAS is strongly related to T cell-mediated inflammation—renal T cells not only express type 1 angiotensin II receptor but also synthesize angiotensin II [88–90]. Thus, RAAS activation in AKI-heart cross talk may amplify the deleterious effect on cardiac inflammation by a T cell-mediated mechanism.
Neurologic manifestations are well known complications of AKI. The blood-brain barrier (BBB) interacts with circulating immune cells, leading to loss of the endothelial lining [91], and AKI-related systemic inflammation can induce BBB disruption [92]. Ischemic AKI can increase levels of soluble inflammatory proteins in the brain, which is associated with abnormal neuropsychiatric behaviors [93,94]. As an additional key mechanism, neurohormonal changes have been reported to contribute to kidney-brain crosstalk [95]. Renocerebral sympathetic reflex accompanied by both renal and cerebral RAAS activation play a crucial role in AKI-induced brain inflammation [96].
Gut microbiota dysbiosis is related to kidney-gut crosstalk [97]. AKI was linked to altered gut microbial populations at the genus and family levels [98]. The bidirectional relationship between the gut microbiota and AKI was discovered using GF mice [9]. The GF mice exhibited worse renal outcomes following IRI and had more NK T cells in the kidney. Reconstitution of the gut microbiota by stool transfer from WT mice before IRI alleviated their susceptibility to IRI [9]. Also, transfer of stool from WT post-IRI mice led to exacerbation of renal injury in GF mice [99]. The gut microbiota exerts immunomodulatory effects via its metabolites [100,101]. These include short-chain fatty acids (SCFAs), which have an anti-inflammatory effect by modulating immune cell function. SCFAs promote Treg proliferation in the intestine and reduce dendritic cell accumulation in the kidney [102,103]. A lack of a microbiota in GF mice affects T cell differentiation and function due to the absence of SCFAs [104].
NOVEL TECHNIQUES FOR STUDYING T CELLS IN AKI
Opportunities to apply novel techniques to improve understanding of kidney T cells in AKI pathophysiology are increasing. Below we briefly discuss recent notable technological advances, including spectral flow cytometry, scRNA-seq, and spatial transcriptomics.
Although fluorescence-based flow cytometers are commonly used to study kidney immune cells, their low resolution and limited multiplexing capacity have discouraged more sophisticated analyses. The recently developed spectral cytometers record full emission patterns across the entire wavelength range, whereas conventional flow cytometers detect only the peak emission signals [105]. A spectral unmixing algorithm enables differentiation of fluorophores with similar peak signals and extraction of autofluorescence contributions, allowing highly complex multicolor panels [106]. The combination of spectral flow cytometry and machine learning-based high-dimensional analysis enables exploration of the roles of immune cells in kidney diseases [107,108].
Although microarray or bulk RNA sequencing data provide the average transcriptomic profiles of predominant cell populations, scRNA-seq assesses gene expression at individual cell levels. This enables to exploit changes in specific cell types independently of predefined hierarchies [109,110]. As such, scRNA-seq enables identification of unrecognized cell populations or discovery of candidate markers for previously defined cell populations. In the past few years, many sensitive and accurate scRNA-seq protocols have been developed [109,110].
Spatial transcriptomic sequencing combines the benefits of histology with high throughput RNA sequencing [111]. RNAs released from intact tissue sections are captured by arrayed oligo-dT spots and bar codes with spatial information. Because the major limitation of single-cell based approaches is loss of spatial information within the tissue, integrating data from spatial transcriptomics and scRNA-seq allow mapping of transcriptomically defined cell types onto a tissue [112]. Although the spatial transcriptomic techniques need more improvement due to their limited resolution, further development of spatial transcriptomic techniques can potentially clarify the spatiotemporal fingerprints of immune cells.
NOVEL THERAPEUTIC APPROACHES
Based on the important roles of T cells in AKI, therapeutic approaches targeting T cells have shown promising outcomes. Below we briefly discuss interventions using adoptive cell transfer, stem cell-based therapy, and approaches targeting gut-kidney crosstalk.
T cell-based therapy
Given the anti-inflammatory properties of Tregs and DN T cells, adoptive transfer of these cells has been investigated in preclinical studies. Adoptive transfer of Tregs before AKI attenuated structural and functional renal injury in both IRI and cisplatin AKI models [27,113]. Moreover, Treg transfer after AKI enhanced tubular regeneration and improved renal outcomes during the recovery phase [56]. Adoptive transfer of ex-expanded Tregs by ex vivo rapamycin treatment attenuated tubular injury and reduced expression of fibrosis markers [70]. DN T cell transfer ameliorated renal injury in both IRI and cisplatin AKI models [31,114], but the effect on renal recovery is unknown. Given that clinical trials of Treg adoptive transfer in kidney transplant recipients showed feasibility and a favorable safety profile [115], cell-based therapies show promise for AKI. However, the preparation required, such as cell collection, isolation, and ex vivo expansion, hamper clinical application because the clinical course of AKI is unpredictable in most patients.
Stem cell-based approaches
Despite the promising results from preclinical trials involving direct injection of mesenchymal stem cells (MSCs) [116,117], this approach has been questioned as the studies failed to achieve even modest survival durations for transplanted cells. Therefore, the beneficial effects did not rely on differentiation of the injected cells [116,117]. Rather, transplanted cells appear to promote tissue regeneration by passing signals in exosomes or microparticles to surrounding cells [118]. Although the mechanism of their paracrine effect is unclear, much evidence supports the immunomodulatory properties of stem cells and the critical role of T cells [119]. MSC treatment in mice attenuated renal injury by facilitating Treg trafficking to the injured kidneys in both IRI and cisplatin AKI. And the protective effect was blunted by anti-CD25 antibody-mediated depletion of Tregs [120,121]. MSC treatment after reperfusion in rat IRI reduced the expression of inflammatory cytokines, including IL-1β, TNF-α, and IFN-γ, and increased levels of anti-inflammatory cytokines, including IL-10 and basic fibroblast growth factor [116]. Stem cell-based therapies appear to be safe and promising for AKI [118], and a clinical trial is underway (NCT03015623).
Therapeutic strategies targeting gut-kidney crosstalk
The mechanistic evidence supporting a bidirectional relationship between the gut microbiota and AKI led to interventional studies targeting the gut microbiota. Gut microbiota depletion using broad-spectrum antibiotics (ampicillin, neomycin, vancomycin, and metronidazole) showed a protective effect on AKI, and stool transplantation from untreated mice abolished their renoprotective effect [122]. Importantly, a reduced Th1 and Th17 response and Treg expansion were suggested as the mechanism by which antibiotic microbiota depletion attenuates renal injury [99]. By contrast, GF mice showed deteriorated renal outcomes following IRI [9], and another combination of antibiotics (ampicillin, vancomycin, and levofloxacin) resulted in more severe structural injury [123]. Therefore, selective deletion of deleterious microbiota populations may exert a protective effect on AKI [124].
Because the gut microbiota controls immune cells via metabolites [124], microbiota-derived products were tested in AKI models. SCFA ameliorated IRI and inhibited dendritic cell maturation, thereby reducing CD4+ and CD8+ T cell proliferation [102]. Similarly, a gut microbial D-amino acid product, D-serine, protected against IRI [125], which warrants further investigation.
The microbial taxa responsible for the protective or exacerbating effects on AKI are unknown. Studies of interventions targeting the gut-kidney axis and sequencing analyses of the gut microbiota may lead to the development of AKI-specific prebiotic or probiotic treatment approaches.
CONCLUSIONS
T cells are important in the pathogenesis of ischemic AKI. Recent advances in our understanding of their roles in repair and regeneration have provided insight into the AKI to CKD transition. Thus, immunomodulatory therapies targeting the T cell mediated pathophysiology show promise for improving AKI outcomes. Other types of immune cells such as mononuclear phagocytic cells, neutrophils, B cells, and innate lymphoid cells are also involved in ischemic AKI pathogenesis, although we did not review them here. Future studies using cutting-edge technologies will facilitate the development of precision-directed therapies.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print





