


INTRODUCTION
Rheumatoid arthritis (RA) is a systemic autoimmune disease characterized by synovial inflammation and subsequent joint destruction [1]. Among the various pathologic events occurring in the affected joints, bone destruction is the most clinically relevant feature because it is related to functional impairment and the progression of joint damage among patients with RA in whom the disease is in prolonged remission [2]. Osteoclasts, which are implicated in the development of bony erosions in RA, are specialized bone-resorbing cells regulated by receptor activator of nuclear factor κ-B ligand (RANKL) and macrophage colony-stimulating factor (M-CSF) [3].
Among the various immune cells, CD4+ T cells are known to be involved in the pathogenesis of RA. RA was previously considered to be a purely Th1-mediated disease. This classic paradigm was maintained until 2005, when a distinct lineage of proinflammatory T helper cells, termed Th17 cells, was identified [4]. Th17 cells are characterized by the production of a proinflammatory cytokine, interleukin 17 (IL-17). Th17 cells and IL-17 significantly contribute to the development and progression of RA [5]. IL-17 induces RANKL expression by synovial fibroblasts and osteoblasts to drive bone erosion indirectly (via metalloproteases), enable activated T cells to penetrate the extracellular matrix [6], and activate synovial macrophages to secrete tumor necrosis factor α (TNF-α) and IL-1β, two major osteoclastogenic factors [7]. Synovial macrophages can be differentiated into fully functional bone-resorbing osteoclasts, and Th17-induced differentiation of synovial macrophages into osteoclasts represents an important cellular mechanism in bone destruction associated with RA [8].
Increased oxidative stress and elevated production of TNF-α, IL-1β, and IL-6 affect bone metabolism in the context of obesity through several mechanisms [9]. Proinflammatory cytokines and oxidative stress are both capable of stimulating osteoclast differentiation and activation [10], and reactive oxygen species (ROS) directly stimulate osteoclast formation and activity, leading to bone resorption [11]. Restoring antioxidant defenses by administration of N-acetyl-l-cysteine (NAC), an antioxidant and glutathione precursor, or ascorbate inhibits RANKL-induced osteoclastogenesis [12] and prevents estrogen deficiency-induced bone loss in mice [13]. In contrast, increasing oxidative stress by depleting glutathione using the specific glutathione synthesis inhibitor l-buthionine-(S,R)-sulfoximine (BSO) increases osteoclast differentiation and causes substantial bone loss [13]. Furthermore, NAC supplementation in the diet mitigates bone loss, downregulates Wnt signaling, and decreases serum bone-formation markers in an ethanol-induced oxidative stress mouse model [14]. The benefit of NAC is demonstrated by its ability to prevent the expression of lipopolysaccharide-induced inflammatory mediators (IL-1β, IL-6, and IL-8) in phagocytic cells and gingival fibroblasts during the inflammatory process, suggesting a direct link between the production of cytokines and the generation of ROS [15]. As a direct result of these actions, NAC suppresses osteoclast differentiation, which leads to the prevention of bone resorption [12].
In this study, we assessed whether IL-17 played a crucial role in the process of bone destruction in RA through upregulation of RANKL or direct induction of osteoclast differentiation. Additionally, we evaluated whether NAC suppressed IL-17-induced RANKL production and osteoclastogenesis. Our findings provided important insights into the potential applications of this antioxidant as a new therapeutic option for RA-associated joint destruction.
METHODS
Patients
Informed consent was obtained from all patients, and the experimental protocol was approved by Institutional Review Board for Human Research, Konkuk University Hospital (KUH1010186). Synovial tissues were isolated from eight RA patients (mean age, 63.4 ± 4.6 years; range, 38 to 76) undergoing total knee replacement surgery.
Reagents
Recombinant human RANKL (rhRANKL) and M-CSF were purchased from R&D Systems (Minneapolis, MN, USA). NAC was obtained from Sigma Chemical Co. (St. Louis, MO, USA). Anti-human IL-17, anti-interferon γ (anti-IFN-γ), anti-IL-10, anti-IL-2, anti-IL-1β, anti-TNF-α, anti-IL-6, and anti-RANKL antibodies were purchased from R&D Systems.
Identification of ex vivo peripheral blood mononuclear cells
Peripheral blood mononuclear cells (PBMCs) were prepared from heparinized blood by Ficoll-Hypaque (GE Healthcare, Chicago, IL, USA) density-gradient centrifugation.
Induction of Th17 polarizing conditions
To induce Th17 polarizing conditions, PBMCs were incubated for 48 hours with anti-CD3 (1 μg/mL; BD, Franklin Lakes, NJ, USA), anti-CD28 (1 μg/mL; BD), IL-1β (20 ng/mL), IL-6 (20 ng/mL), IL-23 (20 ng/mL), IFN-γ-neutralizing antibody (2 μg/mL), and IL-4-neutralizing antibody (2 μg/mL). To examine the immunosuppressive effects of NAC, PBMCs were pre-incubated for 1 hour with NAC, and then stimulated as described above.
Expression of RANKL mRNA by real-time polymerase chain reaction
Synovial fibroblasts were stimulated with IL-17. For RANKL signal pathway analysis, the fibroblast-like synoviocyte (FLS) were incubated in the presence or absence of NAC for 3 hours before the addition of IL-17. After incubation for 72 hours, mRNA was extracted using RNAzol B (Biotex Laboratories, Houston, TX, USA) according to the manufacturer’s instructions.
Enzyme-linked immunosorbent assay
Soluble RANKL, IL-17, IFN-γ, and IL-2 levels in the culture supernatants from RA-FLS or PBMCs were measured using sandwich enzyme-linked immunosorbent assay according to the manufacturer’s instructions.
Western blotting analysis
The primary antibody to phospho-mammalian target of rapamycin (mTOR), AMP-activated protein kinase (AMPK), Akt, phospho-c-Jun N-terminal kinase (JNK), phospho-extracellular signal-regulated kinase, or phospho-inhibitor of κB α (IκB-α, Cell Signaling Technology Inc., Danvers, MA, USA) was diluted 1:1,000 in 0.1% Tween 20/1x Tris-buffered saline (TTBS), and incubated overnight at 4°C. The membranes were washed with TTBS, horseradish peroxidase-conjugated secondary antibody was added, and the membranes were incubated for 1 hour at room temperature. After washing with TTBS, the hybridized bands were detected using an ECL detection kit (Amersham Pharmacia, Piscataway, NJ, USA).
Flow cytometric analysis
Cells were stained with combinations of the following mAbs: CD4-PE/Cy7 and CD25-APC (BD). Cells were washed, fixed, permeabilized, and stained to detect intracellular cytokines with mAbs to IL-17, IFN-γ, IL-4, and forkhead box P3 (Foxp3, eBioscience, San Diego, CA, USA). Cells were analyzed on a FACS Calibur flow cytometry system (BD).
Osteoclast formation
As described above, monocytes were added to the IL-17-pretreated FLS with fresh media. Monocytes were co-cultured for 3 weeks in α-minimal essential medium and 10% fetal bovine serum in the presence of 25 ng/mL recombinant human M-CSF. The addition of rhRANKL protein, prepared as described previously [18], was used as a positive control. On day 21, tartrate-resistant acid phosphatase (TRAP)-positive cells were identified using a leukocyte acid phosphatase kit according to the manufacturer’s protocol [19].
RESULTS
NAC reduced IL-17-induced RANKL gene expression and protein production in RA synovial fibroblasts
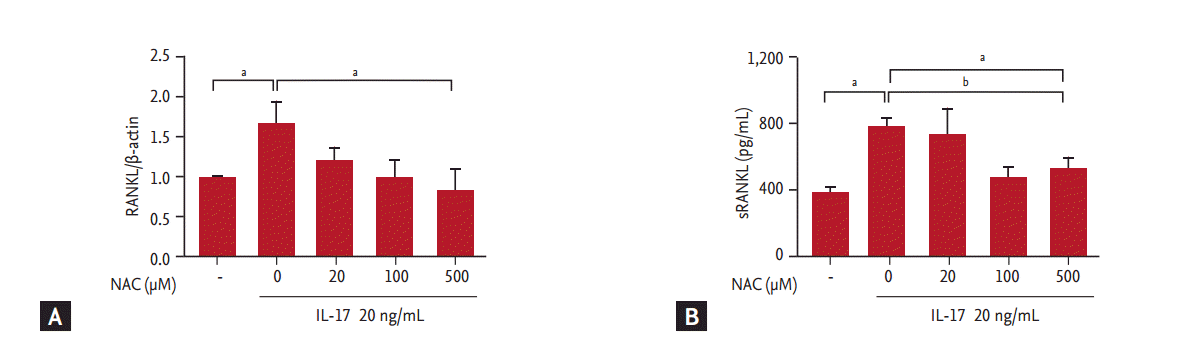
To confirm the inhibitory effects of NAC in IL-17-induced RANKL expression, RA synovial fibroblasts were pre-incubated with NAC for 3 hours. The synovial fibroblasts were then cultured with various concentrations of IL-17 for 72 hours. Our results showed that IL-17 induced RANKL expression and production. Maximal effects were observed at a concentration of 20 ng/mL; therefore, we used 20 ng/mL as an optimal dose (data not shown). NAC reduced the IL-17-induced expression of RANKL mRNA in a concentration-dependent manner (Fig. 1A). NAC also decreased the production of RANKL by synovial fibroblasts, showing a pattern similar to that observed for mRNA expression (Fig. 1B). NAC did not affect the IL-17-induced production of IL-1β, TNF-α, and IL-16 (data not shown). The experimental concentrations of NAC had no cytotoxic or proliferative effects on synovial fibroblasts (data not shown).
Signal pathways involved in the inhibitory effects of NAC in RA synovial fibroblasts
Using RA synovial fibroblasts, we investigated the molecular mechanisms through which NAC modulates IL-17. As shown in Fig. 2, IL-17 enhanced the phosphorylation of mTOR, JNK, and inhibitor of κB α (IκB-α), whereas NAC significantly decreased the IL-17-induced phosphorylation of mTOR, JNK, and its downstream protein IκB-α (p < 0.05 for each).
The regulatory effects of NAC in IL-17-induced osteoclast differentiation
Peripheral blood CD14+ monocytes are osteoclast precursors and can differentiate into TRAP+ multinucleated osteoclasts in the presence of RANKL and M-CSF, representing a traditional culture system for osteoclastogenesis from osteoclast precursors [20-22]. To evaluate the effects of NAC on IL-17-induced osteoclast differentiation, isolated CD14+ monocytes from human peripheral blood were cultured with IL-17 and M-CSF in the absence of RANKL. After 21 days of culture, TRAP+ multinucleated osteoclasts were differentiated. However, preincubation with NAC significantly decreased IL-17-induced osteoclastogenesis in a concentration-dependent manner (Fig. 3A). The mRNA expression of osteoclast markers, such as TRAP, RANK, and calcitonin receptor was also increased with IL-17 stimulation but reduced by NAC treatment (data not shown).
The isolated CD14+ monocytes were cultured with RANKL and M-CSF. After 21 days of culture, TRAP+ multinucleated osteoclasts were differentiated from their precursors in the RANKL and M-CSF culture system. Notably, the osteoclastogenic effect was three times greater than that in the IL-17 and M-CSF culture system. However, preincubation with NAC significantly decreased RANKL-induced osteoclastogenesis (Fig. 3B).
RA-FLS were pretreated with IL-17 and NAC and then were added to a culture system with CD14+ monocytes and M-CSF. When the monocytes were co-cultured with IL-17-pretreated RA-FLS in the absence of RANKL, TRAP+ multinucleated osteoclasts showed greater differentiation than that observed when monocytes were co-cultured with untreated RA-FLS. NAC suppressed the differentiation of osteoclasts induced by IL-17-pretreated FLS (Fig. 3C). The mRNA expression levels of osteoclast markers, such as TRAP, RANKL, and cathepsin K, were increased in co-culture with IL-17-pretreated FLS; however, NAC blocked this effect (data not shown).
When osteoclast precursors were cultured with CD4+ T cells, osteoclastogenesis was decreased because of the antiosteoclastogenic effects of IFN-γ produced by CD4+ T cells. However, Th17 cells have high inflammatory and destructive potential and can induce osteoclastogenesis [23]. When isolated monocytes were co-cultured with Th17 cells and M-CSF in the absence of RANKL, TRAP+ multinucleated cells showed increased differentiation, albeit without statistical significance. NAC suppressed the differentiation of osteoclasts induced by untreated T cells and Th17 cells (Fig. 3D).
Effects of NAC on Th17 cell differentiation
PBMCs were isolated from healthy individuals and cultured under Th17-polarizing conditions in the presence of NAC (Fig. 4A). NAC significantly reduced the percentage of IL-17+/CD4+ T cells. However, NAC did not affect the differentiation of the cells into CD25+Foxp3+CD4+ T cells, IFN-γ+CD4+ T cells, or IL-4+CD4+ T cells (Fig. 4B). NAC also reduced the secretion of IL-17 and RANKL into the cultured medium (Fig. 4C).
DISCUSSION
In RA, oxidative stress is an important mechanism causing destructive proliferative synovitis [11]. Moreover, overproduction of TNF-α increases ROS production, resulting in inflammation and tissue destruction. RANKL induces the expression of nitric oxide synthase, and NAC attenuates RANKL-induced ROS production and osteoclast differentiation in bone marrow monocyte-macrophage lineage cells [12]. Additionally, osteoclasts are activated by ROS to enhance bone resorption [24]; however, few studies have investigated the role of ROS in the differentiation of macrophages and monocytes into osteoclasts. Sequential processes are involved in osteoclast differentiation and function, including differentiation from osteoclast precursors, cellular fusion to multinucleated osteoclasts, activation of multinucleated osteoclasts, and sustained survival of the osteoclasts [25,26]. M-CSF, RANKL, TNF-α, IL-17, and IL-1β are the primary molecules involved in osteoclastogenesis, although various other cytokines and chemokines are also involved in this process in RA [26]. When osteoclast precursors were cultured with RA-FLS, the cells differentiated into mature osteoclasts more efficiently than when the cells were cultured alone [27]. Synovial fibroblasts are the primary cells involved in RA; these cells have many receptors and produce various inflammatory mediators, facilitating cross-talk with other immune cells and cytokines.
NAC is a thiolic antioxidant produced by the body and serves as a precursor of glutathione synthesis [28]. The efficacy of NAC has been investigated in clinical trials and experimental models of various respiratory conditions, such as chronic obstructive pulmonary disease, cirrhosis, portal hypertension, and acetaminophen poisoning [29]. NAC decreases the cytogenetic damage produced by exposure to cigarette smoke, whereas the intravenous or intra-arterial application of NAC is protective against the side effects of chemotherapy [30]. Additionally, as an antioxidant, NAC has inhibitory effects on osteoclastogenesis [12]. Accordingly, in this study, we investigated the antioxidant effects of NAC in IL-17-induced osteoclastogenesis and Th17 differentiation in RA.
We found that NAC reduced IL-17-induced RANKL gene expression and protein synthesis in RA synovial fibroblasts. IL-17 is a major cytokine involved in osteoclast differentiation. In our recent study, Th17 cytokines, such as IL-17, IL-21, and IL-22, were found to induce RANKL expression in RA synovial fibroblasts [23]. When peripheral blood monocytes are cultured with M-CSF and the Th17 cytokines in the absence of RANKL, the three Th17 cytokines significantly induce osteoclast differentiation from monocytes. Indeed, although IL-17 is a strong inducer of osteoclastogenesis, other Th17 cytokines, such as IL-21 and IL-22, also have osteoclastic effects, including effects that are different from those of IL-17. In RA treatment, the clinical efficacy and safety of anti-IL-17 monoclonal antibodies, such as secukinumab and ixekizumab, have been under clinical investigation [31]. However, in a phase II study of secukinumab for patients with RA, inadequate responses were observed. Despite this, phase III studies are still underway, and IL-17 is still promising target for RA treatment [32]. Anti-IL-17 monoclonal antibodies are developed by biologic methods; however, NAC is a chemical compound that has many advantages, including low cost, capacity for oral administration, short half-life, and low adverse effects. Similar benefits for the Janus kinase inhibitor tofacitinib have increased the use of this drug [33], although NAC would be more likely to have applications in enhancing the efficacy of other biologic therapies for preventing joint destruction.
In terms of signal transduction pathways, the inhibitory effects of NAC in IL-17-induced activation of RA synovial fibroblasts are known to be mediated through the mTOR/JNK/nuclear factor κB (NF-κB) pathway. IL-17 activates mTOR, and NAC reduces IL-17-induced mTOR activation. Additionally, NAC activates AMPK, which functions to counteract the mTOR signal. In RA, inhibition of mTOR complex 1 (mTORC1) alters the ability of IL17 to induce mTORC1-dependent proliferation of RA synovial fibroblasts [34]. Interestingly, mTORC1 activity is increased in osteoclasts from patients with RA and in arthritic transgenic mice [35], and inhibition of mTOR results in downregulation of extracellular matrix digestive enzymes and induction of apoptosis in osteoclasts [35]. NAC inhibits nuclear translocation of NF-κB in RA synovial fibroblasts [36] and keratinocytes [37]. Moreover, Sakurada et al. [36] suggested that the inhibition of NF-κB nuclear translocation by NAC subsequently suppresses the induction of several cytokines and the expression of intracellular adhesion molecule-1.
In our study, we found that IL-17 directly induced osteoclast differentiation and indirectly induced osteoclastogenesis through co-culture of osteoclast precursors with RA synovial fibroblasts pre-exposed to IL-17 and Th17 cells. Additionally, NAC suppressed IL-17-induced osteoclastogenesis and osteoclast differentiation from co-cultures of their precursors with IL-17-pretreated synovial fibroblasts and Th17 cells. Based on this result, IL-17 affects many stages of osteoclastogenesis, and NAC regulates IL-17-induced osteoclastogenesis.
Finally, we found that NAC regulated Th17 cell differentiation but did not affect the differentiation of regulator T cells or Th1 cells. This result suggested that NAC may modulate Th17 cell differentiation and cytokine production; thereby, attenuating Th17 differentiation, which is crucial for the pathogenesis of synovial inflammation and bony destruction.
In conclusion, our results demonstrated that NAC regulated IL-17-induced RANKL expression in RA synovial fibroblasts and osteoclastogenesis from precursor cells. NAC also suppressed IL-17-activated synovial fibroblasts and Th17 cell-induced osteoclastogenesis. Although NAC has limitations in the control of inflammation and tissue damage, NAC could be administered as a supplemental therapeutic for prevention of inflammation and bone destruction in RA.
KEY MESSAGE
1. Stimulation of rheumatoid arthritis (RA) synovial fibroblasts with interleukin 17 (IL-17) promoted receptor activator of nuclear factor κ-B ligand (RANKL) production.
2. N-acetyl-l-cysteine (NAC) blocked IL-17-induced RANKL production in a concentration-dependent manner.
3. NAC decreased IL-17-induced activation of mammalian target of rapamycin/c-Jun N-terminal kinase/inhibitor of κB signaling.
4. Under Th17-polarizing conditions, NAC decreased Th17 cell differentiation.
5. NAC could be a supplementary therapeutic option for inflammatory processes in RA.



 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print





