


To the Editor,
Human pubertal development and fertility are regulated mainly by luteinizing hormone (LH) and follicle stimulating hormone (FSH). LH and FSH, secreted from common gonadotroph cells, exist as heterodimers of a common ╬▒-subunit and a specific ╬▓-subunit, each encoded by a separate gene. Mutations in gonadotropin genes are very rare. This might be explained by selection due to the direct effects of such mutations on fertility and reproduction. Isolated deficiency of LH due to mutations in the ╬▓-subunit gene is a very rare cause of hypogonadotropic hypogonadism. Only four homozygous inactivating mutations have been reported to date, in six men and two women [1-4]. Recently, a compound heterozygous mutation in the LH ╬▓-subunit gene was described in a man with isolated LH deficiency [5]. Herein, we describe a young man with delayed puberty induced by a selective deficiency in LH. The patient had a novel mutation in the LH ╬▓-subunit gene. To our knowledge, this is the first case of hypogonadotropic hypogonadism due to a mutation in the LH ╬▓-subunit gene diagnosed in Asia.
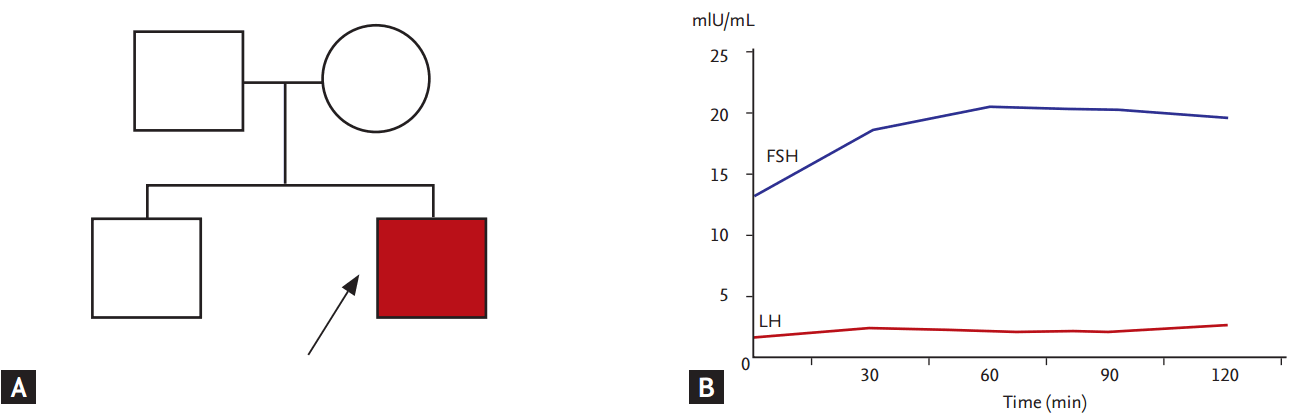
A 19-year-old man was referred for evaluation of delayed puberty. He was 184 cm tall, weighed 80 kg, and had an arm span of 188 cm. He had a prepubertal physique, bilateral gynecomastia, and a juvenile voice. He had sparse axillary and pubic hairs (Tanner stage II). The testes were small and soft, 9/4.5 mL in volume. The penile length was 3.5 cm. No family members were available for genetic testing, but he had no family history of infertility or delayed puberty and his 24-year-old brother, the only sibling, was healthy (Fig. 1A). The results of the initial laboratory tests were as follows: a low testosterone level (0.2 ng/mL; reference range, 2.7 to 10.7), a low LH level (0.6 mIU/mL; reference range, 2.0 to 12.0), and an elevated FSH level (15.9 mIU/mL; reference range, 1.0 to 12.0). Serum prolactin, free thyroxine, thyrotropin (TSH), basal 8:00 AM cortisol and adrenocorticotropin concentrations were normal. Ninety minutes after intravenous administration of gonadotropin-releasing hormone (100 ╬╝g), the FSH level increased from 13.2 to 20.5 mIU/mL; however, no significant increase in the LH level (1.6 mIU/mL at 0 minute, 2.0 mIU/mL at 90 minutes) was detected (Fig. 1B). Magnetic resonance imaging of the sellar region showed no abnormalities. The patientŌĆÖs karyotype was 46,XY. From these clinical and biochemical characteristics, a diagnosis of hypogonadism due to an isolated LH deficiency was made. Treatment with intramuscular testosterone enanthate (100 mg every 2 weeks) was initiated with gradual increment in dose to 200 mg every 3 weeks over 1-year period. Testosterone therapy induced physical changes of puberty and penile growth.
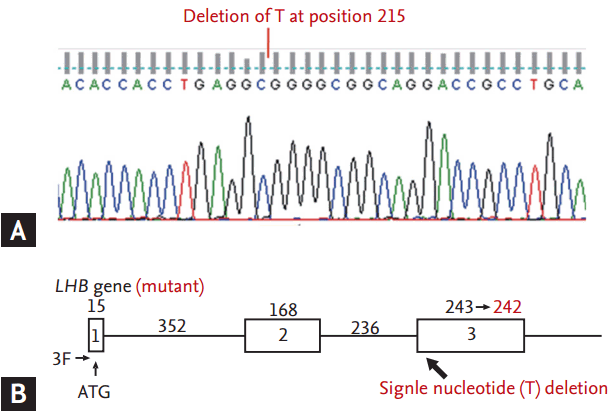
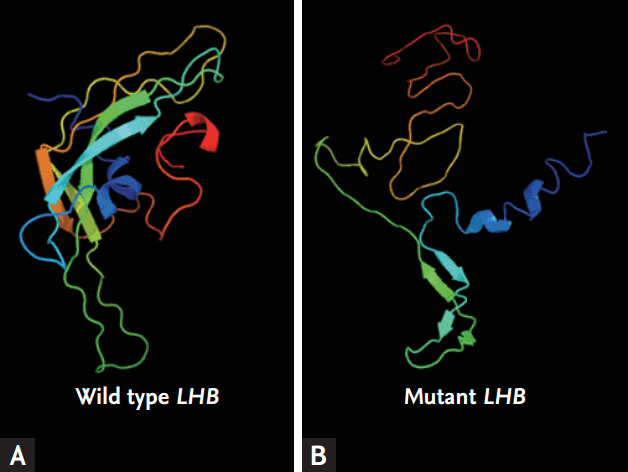
The three exons and two introns of the human LH ╬▓-subunit gene encode a 24-amino acid leader peptide and a 121-amino acid mature protein. For mutation analysis, genomic DNA was extracted from the peripheral blood leukocytes of the patient. All three exons and two introns of the LH ╬▓-subunit gene were amplified by polymerase chain reaction using a thermal cycler. The 5ŌĆÖ primer was 5ŌĆÖ-GCACCAAGGATGGAGATGCTCCAGGTAAGACTACAGGGCCCCTGGGCA-3ŌĆÖ (LHF), and the 3ŌĆÖ primer was 5ŌĆÖ-GAGCCCACAGAAAGACACCTCCAGAGTGCGGATTGAGAA-3ŌĆÖ (LHR). The amplified products (1,200 bp) were sequenced in both the sense and antisense directions using the LHF or LHR primers. The sequences were compared with the reference sequences, and the GenBank accession numbers were X00264.1 and NM000894.2. The numbering of the nucleotide positions was performed according to the translation start site; the intronic sequences are excluded. Analysis of the sequence revealed a point deletion of the thymine nucleotide at position 215 [215 del T (p.Leu72Arg)] in exon 3, which would cause a frameshift and premature termination at codon 128 (Fig. 2). A hypothetical model of the mutated LH ╬▓-subunit of this patient was constructed using the Phyre2 (Protein Homology/analogY Recognition Engine version 2.0, Structural Bioinformatics Group Imperial College, London, England) program (Fig. 3).
In men, LH induces testosterone synthesis and secretion from Leydig cells, and FSH stimulates seminiferous tubule development and regulates spermatogenesis in concert with testosterone. Because testicular testosterone production is crucially dependent on LH stimulation, isolated LH deficiency manifests with delayed puberty, eunuchoid body proportions and arrested spermatogenesis because of low testosterone levels. The patient was phenotypically male at birth, with descended testes, showing that pituitary LH becomes important only after birth and is critical to the induction of puberty.
Selective deficiency of LH due to inactivating mutations in the ╬▓-subunit gene is a very rare cause of hypogonadotropic hypogonadism. This patient had a novel mutation in the coding sequence of the LH ╬▓-subunit gene. Homozygous point deletion of the thymine nucleotide at position 215 [215 del T (p.Leu72Arg)] in exon 3, observed in this patient, is predicted to result in a frameshift and premature termination at codon 128, suggesting complete loss of function of the protein. Homozygous inactivating mutations in the LH ╬▓-subunit gene have been found to date in only six men and two women with delayed puberty and isolated LH deficiency. The genotypic abnormalities were as follows: a missense mutation that prevented receptor binding [1], a missense mutation in exon 2 that disrupted a vital cysteine knot motif and abrogated the heterodimerization and secretion of LH [2], a nine-base deletion in exon 2 that impaired the cysteine knot-folding motif [3]; and a G-to-C substitution in intron 2 that disrupted splicing, leading to abnormal processing of the ╬▓-subunit mRNA [4]. Recently, a compound heterozygous mutation in the LH ╬▓-subunit gene has been described in a man with isolated LH deficiencyŌĆöa 12-base deletion in exon 2 and a G-to-T substitution in intron 2 present in a compound heterozygous state that impairs the maturation of the ╬▓-subunit peptide and that disrupts splicing of the mRNA, respectively [5]. The limitation of our study includes a lack of genetic testing of family members and functional analysis of mutant ╬▓-subunit.
Gonadotropin and TSH ╬▓-subunit mutations are extremely rare. Interestingly, inactivating mutations in the gene encoding the common ╬▒-subunit of these glycoprotein hormones have not been reported, likely because such impairments in human chorionic gonadotropin (hCG), TSH and gonadotropins cause lethality to the fetus. In addition to the very rare mutations reported, there are more common polymorphisms, particularly in the LH ╬▒-subunit gene, that might affect hormonal function. They may also alter the immunoreactivity of the hormone and results of hormone assays.
Regarding the treatment of hypogonadotropic hypogonadism, testosterone replacement is required to induce and maintain primary and secondary sexual functions. Because testosterone therapy may suppress spermatogenesis, the steroid should be discontinued before hCG treatment is initiated to induce spermatogenesis. However, infertility and very low levels of spermatogenesis may persist in affected men, despite long-term exposure to hCG, suggesting that the absence of perinatal exposure to LH alters Leydig cell proliferation and maturation.
In conclusion, we report a case of a young man with delayed puberty associated with a selective deficiency of LH due to a novel mutation of homozygous point deletion of the thymine nucleotide at position 215 [215 del T (p.Leu72Arg)] in exon 3 of the LH ╬▓-subunit gene. Although LH ╬▓-subunit mutations are very rare and not readily detected by clinicians, they should be considered in patients with delayed puberty and selective LH deficiency.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print





