


INTRODUCTION
Inflammatory bowel disease (IBD) is a chronic disease characterized by immune activation and inflammation of the gastrointestinal tract with etiology that is not yet clearly understood. Although the primary pathology in ulcerative colitis (UC) and Crohnâs disease (CD) is in the gastrointestinal system (GIS), some extra-intestinal involvements may be encountered, including in the skin (erythema nodosum, pyoderma gangrenosum), eyes (uveitis, episcleritis), liver (pericholangitis, fatty liver), and joints (peripheral and axial arthropathy) [1]. For the first time in 1976, Kraft et al. [2] found evidence of pulmonary involvement in IBD. Subsequent to this publication, diverse pulmonary involvements that might be encountered in IBD have been identified in many cases and in small case series; these involvements include inflammatory tracheal stenosis, peribronchiolitis, interstitial pneumonia, Wegener granulomatosis, alveolitis, serositis, bronchitis, and bronchiectasis [3-5].
The fact that both the lungs and the GIS embryologically arise from the primitive gut suggests, theoretically, that intestinal changes may also occur in the lungs. Another theory for pulmonary involvements in IBD is that the impaired intestinal barrier allows for the entrance of more antigens and bacteria into the organism, increasing both local and systemic inflammatory response in the body [2].
Over the course of the development of IBD, many proinflammatory cytokines, primarily tumor necrosis factor α (TNF-α), interleukin (IL)-1β, and IL-6, are produced by macrophages and lymphocytes found in the inflamed intestinal mucosa. These proinflammatory cytokines play a critical role in the maintenance of the inflammatory response [6]. Among these cytokines, the efficacy of TNF-α has been largely demonstrated and forms the basis of a biological treatment model for IBD that has become a standard therapy in recent years. Treatment of the disease is possible with the inhibition of TNF-α production in the inflamed mucosa [7]. Among angiogenesis markers in IBD, vascular endothelial growth factor (VEGF) is the most frequently defined factor. Human studies have found that VEGF levels rise and fall along with disease activity. VEGF increases with activation of IBD and decreases with the administration of steroids and infliximab [8]. In addition, VEGF was found to be higher in rats with alveolar hemorrhage [9].
Pulmonary involvement in IBD is usually asymptomatic. The most common symptoms include coughing, sputum, and dyspnea [10,11]. It has been reported that impaired respiratory function testing (RFT) in asymptomatic IBD patients is in line with disease activity [12]. Findings on chest X-ray and conventional computed tomography are non-specific for pulmonary involvement [5,10]. These findings are usually detected in IBD patients through scanning and RFT. However, although physiological and radiological outcomes have been demonstrated, there is no study exposing pulmonary tissue pathology based on histopathological examination [13]. In clinical studies, it is neither practical nor ethical to histopathologically demonstrate pulmonary involvement in IBD. Therefore, an experimental model of colitis is required.
The present study aimed to histopathologically examine pulmonary involvement in experimentally induced colitis and to explore VEGF and TNF-α concentrations in pulmonary tissue in order to determine the role of proinflammatory cytokines and angiogenesis in the etiopathogenesis of IBD.
METHODS
Experimental animals
The animals involved in the study were maintained and used in accordance with the Animal Welfare Act and the Guide for the Care and Use of Laboratory animals prepared by the Suleyman Demirel University Animal Ethical Committee. The animals were housed in a temperature-controlled room at a steady temperature of 24°C, with light from 8:00 AM to 8:00 PM and free access to water. In the study, 24 male Wistar Albino rats at the age of 18 weeks and weighing 200 to 250 g were used. They were randomly distributed into four groups.
Study protocol
Four groups, each of which included six animals, were formed. Acute colitis was induced in each of two groups using dextran sulphate sodium (DSS) or trinitrobenzene sulfonic acid (TNBS) [14] and the remaining two groups were considered to be control groups for each model of colitis.
Induction of colitis
Induction of colitis with TNBS
The most commonly used model to investigate the pathogenesis of IBD and develop new anti-inflammatory strategies is the TNBS model, in which TNBS is supplied in an ethanol solution. Because ethanol splits the epithelial layer, it allows the lamina propria to be exposed to bacterial components. Colonic damage is created by the intrarectal application of barrier breaker ethanol and hapten mixture [14].
Under intraperitoneal ketamine (80 mg/kg) + xylazine (10 mg/kg) anesthesia, a polyurethane nutritional cannula with an external diameter of 2 mm was inserted into the rats, which had been deprived of food for 24 hours, through the anus and proceeded in the way the tip would be in the proximal 8 cm of the anal wedge. In order to induce acute colitis, 15 mg TNBS was dissolved in 0.25 mL 50% ethanol and instilled into the colon through the cannula. The TNBS-ethanol solution was then completely removed from the cannula by injecting 0.5 mL air. After the instillation, the animals were kept upside-down for a few minutes to prevent the instilled solution from flowing out of the colon [15].
Induction of colitis with DSS
It has been found in mice, hamsters and rats that acute colitis will develop at about the third day when 3% to 5% DSS is added to the animalsâ drinking water every day for a week. This experimental model has been used in many studies [14]. Water given after administering 3% to 5% DSS for 5 to 7 days results in serious intestinal damage through the complete depletion of the colon crypt epithelium and relatively slow regeneration.
An experimental model of colitis was created by adding 3% to 5% DSS to tap water and giving this solution to the animals as drinking water for a week [16].
Macroscopic evaluation of colitis
Evaluation of colitis was performed by an observer who was blind to the treatments that the animals received. Ten centimeters of distal colon was removed in each animal and longitudinally dissected. The colon sections were mildly washed with normal saline, and fecal remnants were removed. Macroscopic inflammation scoring was performed according to the clinical appearance of the colon using the Wallace score. This score rates macroscopic colon lesions on a scale from 0 to 10 based on criteria reflecting inflammation, such as hyperemia, thickening of the bowel, and the extent of ulceration [17].
Histological examination of the lungs
Preparations for histopathological examination of pulmonary samples obtained from sacrificed rats were stained with H&E, and the lesions were examined under a light microscope (Ã200) [18].
Biochemical analyses of tissues
Pulmonary tissue concentrations of VEGF and TNF-α were measured by enzyme-linked immunosorbent assay method (BIOSOURCE, Invitrogen Immunoassay kit, Invitrogen, Carlsbad, CA, USA). Concentrations of VEGF and TNF-α were presented in pictograms. Total protein concentration was determined by the Lowry method[19].
Statistical analysis
A database was formed with the study data using the SPSS version 15.0 (SPSS Inc., Chicago, IL, USA). Data were summarized by descriptive statistics. The Mann-Whitney U test was used and results were presented as mean ± standard deviation, and p < 0.05 was considered statistically significant.
RESULTS
Colon tissue concentrations of VEGF and TNF-α in TNBS-colitis, in DSS-colitis and control group
VEGF concentration was not statistically significantly different either between the TNBS colitis group and its own control group or between the DSS colitis group and its own control group (p = 0.589 and p = 1.000, respectively). TNF-α concentration was statistically significantly higher both in the TNBS colitis group and the DSS colitis group as compared to the control groups (p = 0.009 and p = 0.040, respectively) (Table 1).
There was not any statistically significant difference between the animals with TNBS and DSS of colitis in terms of colon tissue VEGF and TNF-α concentrations (p = 0.470 and p = 0.140, respectively). There was no significant difference between the TNBS control and the DSS control groups in various variables such as VEGF levels (p = 0.147) and TNF-α levels (p = 0.153) in colon tissue.
Histopathological changes and VEGF and TNF-α concentrations in the pulmonary tissue in TNBS colitis, DSS colitis, and control group
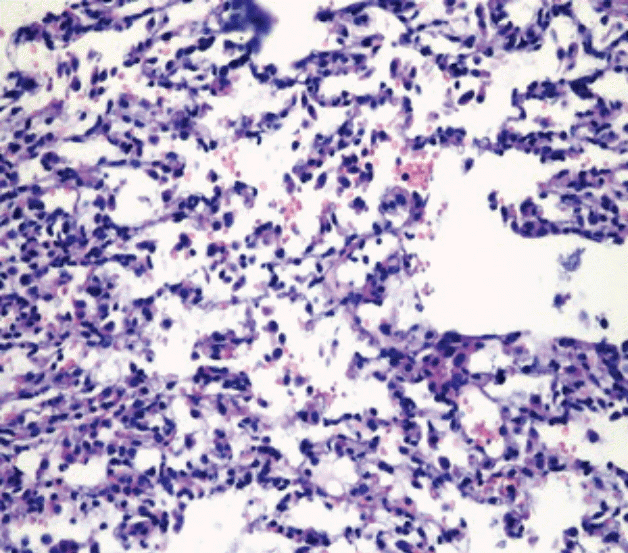
Extensive alveolar hemorrhage was observed in the pulmonary tissues of five animals (83.3%) in the TNBS colitis group and five animals (83.3 %) in the DSS colitis group (Fig. 3), whereas alveolar hemorrhage was detected in the pulmonary tissue of only one animal (17%) in the control arm of each of the colitis groups. The number of animals that had alveolar hemorrhage was significantly higher in the TNBS colitis and DSS colitis groups as compared to their own control groups (p = 0.015 and p = 0.015, respectively). Pulmonary tissues VEGF and TNF-α concentrations were significantly increased in the TNBS colitis and DSS colitis groups compared to their own control groups (p = 0.002 and p = 0.004, respectively; and p = 0.002 and p = 0.002, respectively) (Table 2).
There was no statistically significant difference between the animals with TNBS and DSS of colitis in terms of pulmonary tissue VEGF and TNF-α concentrations (p = 0.065 and p = 0.093, respectively). There was no significant difference between the two control groups of separate colitis models in terms of VEGF levels (p = 0.070) and TNF-α levels (p = 0.465) in pulmonary tissue.
DISCUSSION
The histopathological examination of pulmonary tissues of the animals with colitis found extensive alveolar hemorrhage, demonstrating pulmonary involvement in IBD. It was also observed that pulmonary tissue concentrations of VEGF and TNF-α increased in these animals. Our results indicate that the inflammatory response caused by proinflammatory cytokines (TNF-α), as well as a VEGF-associated increase in alveolar epithelial permeability, may play a role in the etiopathogenesis of IBD-associated pulmonary involvement.
Many proinflammatory cytokines, primarily TNF-α, IL-1, and IL-6, are produced by macrophages and lymphocytes in the inflamed intestinal mucosa over the course of development of IBD, and these proinflammatory cytokines play a critical role in the maintenance of the inflammatory response [6].
Although recent clinical studies have revealed that the prevalence of IBD-associated pulmonary involvement is higher than assumed, information on the etiopathogenesis of IBD and pulmonary involvement in IBD is quite limited [20]. Latent pulmonary involvement has been identified in the RFT, bronchoalveolar lavage fluid and induced sputum samples of a group of asymptomatic patients that were being followed for IBD [10,13,21]. However, the necessity of using invasive methods for histopathological demonstration of pulmonary involvement in IBD patients means that there is still inadequate information available on this subject.
There are two main experimental models of colitis in the literature: TNBS colitis and DSS colitis [14]. We aimed to perform a macroscopic and histopathological examination of potential pathologies in the pulmonary tissue of animals with colitis induced by both models. TNBS colitis, which is often used in studies on the physiopathology of IBD, usually reflects CD, because mucosal inflammation results from Th1 response, which appears due to the production of proinflammatory cytokines [22]. No toxic effect of DSS on the lungs has been reported. On the contrary, it has been reported that DSS prevents pulmonary capillary pressure changes, and DSS has even been used in open heart surgery to prevent lung injury in recent years, with successful results [23,24].
The present study started from the hypothesis that if similar pulmonary pathologies were to occur in both models of colitis (TNBS and DSS), the primary reason for the pathology would be the colitis rather than the agent used to induce colitis. Pulmonary tissue findings in two different models of colitis were compared with those of a healthy control group. Alveolar hemorrhage was detected on histopathological examination of the pulmonary tissue in the groups with TNBS and DSS colitis. Pulmonary tissue VEGF concentration was significantly higher in both the TNBS and DSS colitis groups compared to the control groups. In an animal study, lungs of transgenic rats in which VEGF expression had been enhanced were histopathologically examined; alveolar hemorrhage, hemosiderosis, and signs of inflammation that were not present in the control group were detected [9]. Fifty percent of transgenic rats with enhanced VEGF expression had died in 2 weeks. The high mortality rate suggested that it might have resulted from alveolar hemorrhage and respiratory distress syndrome due to enhanced VEGF, which probably increases vascular permeability [9]. In the present study, histopathological demonstration of alveolar hemorrhage in the colitis groups and accompanying enhanced VEGF suggest that alveolar permeability is increased by VEGF, resulting in alveolar hemorrhage. Thus, demonstration of a significant increase in alveolar permeability by technetium-99m diethylene triamine penta-acetic acid scintigraphy in patients with active disease among 31 UC patients without respiratory complaints, as compared to a control group, corroborates this hypothesis [25]. However, the increase in VEGF in this case may be due to lung involvement.
Although the present study suggested a VEGF-associated increase in alveolar epithelial permeability, the exact mechanism needs to be clarified in the future.
TNF-α, which is secreted by macrophages over the course of IBD, is an important proinflammatory cytokine, and various studies have shown that it plays a critical role in IBD [6,26]. In addition to the maintenance of inflammation, it is known that TNF-α can either directly or indirectly trigger angiogenesis [8]. In the present study, a significant increase in TNF-α concentrations measured in pulmonary and colon tissues in both models of colitis, as compared to the control group, indicates that increased TNF-α might lead not only to colon inflammation, but also to the development of pulmonary pathology and inflammatory response due to proinflammatory cytokines (TNF-α). The angiogenic response triggered by TNF-α might have also played a role in the etiopathogenesis of histopathological changes in the lungs. VEGF-associated alveolar hemorrhage probably begins simultaneously with inflammation in colon tissues. Our results suggest that alveolar hemorrhage associated with increased TNF-α and VEGF in lung tissue of rats with induced colitis can begin simultaneously with inflammation in colon tissue. Because there are no studies in the literature investigating the role of inflammatory cytokines such as TNF-α and VEGF in lung tissues in the experimental colitis model, our study is the first study on the timing of the relationship between lung pathology and colitis.
IBD is a chronic inflammatory disease that courses with activations and remissions. The fact that the present study was carried out in a colitis model created for only seven days is one of its most important limitations. Verification of the findings of the present study in a longer-term chronic colitis model would complement our results. Histological changes in pulmonary tissues and inflammatory cytokines that play a role in inflammation may be different in the acute and chronic colitis periods. It is particularly important to investigate whether the process that begins with alveolar hemorrhage due to TNF-α-associated angiogenesis and increased VEGF in the acute colitis period continues with alveolar hemosiderosis and development of fibrosis in the chronic period.
In conclusion, the present study demonstrated that significant and serious histopathological changes that are directly associated with colitis occur in the lungs in IBD. Histopathological examination of the lungs revealed alveolar hemorrhage in the IBD models created with either TNBS or DSS. Pulmonary involvement probably begins simultaneously with the inflammation in the colon. TNF-α plays an important role in the development of pathologies in both organs. Increased pulmonary VEGF and accompanying alveolar hemorrhage suggest that a pathological process, which is induced by increased alveolar permeability and begins with alveolar hemorrhage, is in question.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print





