Metformin and its therapeutic applications in autoimmune inflammatory rheumatic disease
Article information

Abstract
Metformin is a first-line therapeutic agent for type 2 diabetes. Apart from its glucose-lowering effect, metformin is attracting interest regarding possible therapeutic benefits in various other conditions. As metformin regulates cell metabolism, proliferation, growth, and autophagy, it may also modulate immune cell functions. Given that metformin acts on multiple intracellular signaling pathways, including adenosine monophosphate (AMP)-activated protein kinase (AMPK) activation, and that AMPK and its downstream intracellular signaling control the activation and differentiation of T and B cells and inflammatory responses, metformin may exert immunomodulatory and anti-inflammatory effects. The efficacy of metformin has been investigated in preclinical and clinical studies on rheumatoid arthritis, osteoarthritis, systemic lupus erythematosus, Sjögren’s syndrome, scleroderma, ankylosing spondylitis, and gout. In this review, we discuss the potential mechanisms through which metformin exerts its therapeutic effects in these diseases, focusing particularly on rheumatoid arthritis and osteoarthritis.
INTRODUCTION
Metformin, a first-line therapeutic agent for the treatment of type 2 diabetes, is a biguanide synthetically derived from glucose-lowering herbal medicines. Although its efficacy and safety in type 2 diabetes have been known for decades, the molecular mechanisms of metformin remain under investigation. Studies have found that multiple modes of action are involved in the glucose-lowering effect of metformin [1]. Metformin acts on adenosine monophosphate (AMP)-activated protein kinase (AMPK)-dependent and -independent mechanisms to reduce hepatic glucose production and increase peripheral glucose utilization. AMPK activation of hepatocytes is induced by metformin via reduced cellular energy or upstream AMPK kinase, leading to downregulation of gluconeogenic gene transcription [2,3]. Metformin targets mitochondrial respiratory chain complex 1 to decrease adenosine triphosphate (ATP) and increase the AMP/ATP ratio, and is thereby linked to AMPK activation [4–6]. AMPK-independent mechanisms are also thought to play a role in the regulatory action of metformin on hepatic gluconeogenesis, such as the inhibition of gluconeogenic enzymes, suppression of glucagon signaling, and alteration of a cellular redox state by direct inhibition of the mitochondrial enzymes involved in electron transport shuttle [7–10].
AMPK is a regulator of cellular metabolism that senses cellular energy status. AMPK is a serine/threonine kinase that forms a complex consisting of α, β, and γ subunits [11]. AMPK is activated when cellular AMP binds to the AMPK-γ subunit, leading to phosphorylation of the catalytic AMPK-α subunit. In the case of cellular energy depletion, activated AMPK promotes ATP-producing catabolic pathways (e.g., glycolysis and fatty acid oxidation) and suppresses ATP-consuming anabolic pathways (e.g., gluconeogenesis and lipogenesis), to regulate cellular glucose and lipid metabolism and maintain the energy balance [12,13]. In addition to metabolic regulation, AMPK can also control cell growth and autophagy [14]. The molecular mechanisms underlying the cell growth-regulating action of AMPK include inhibition of the mammalian target of rapamycin complex 1 (mTORC1), which is required for rapid cell proliferation, and activation of tumor suppressor and cell cycle inhibitor proteins [14]. AMPK signaling acts on multiple intracellular pathways, molecules, and transcription factors; therefore, AMPK-activating drugs, such as metformin, may also have beneficial effects in various conditions other than type 2 diabetes and metabolic syndrome.
AMPK and downstream pathways can modulate immune cell function. T cells undergo metabolic reprogramming to adapt to increased energy demands for activation and differentiation; AMPK plays a major role in this process [15]. mTOR, a downstream molecule of AMPK, determines the fate of T cells, i.e., whether they differentiate into effector or regulatory T (Treg) cells [16]. CD4+ T cells require mTORC1 signaling to differentiate into Th1 and Th17 cells, and mTORC2 signaling to differentiate into Th2 cells, while CD4+ T cells lacking both mTORC1 and mTORC2 signaling differentiate into Treg cells [16,17]. The association between the selective expression of mTOR and differentiation of CD4+ T cells is mediated by differential expression of transcription factors, including signal transducer and activator of transcription (STAT). AMPK can directly or indirectly suppress JAK-STAT pathway to exert anti-inflammatory and immunomodulatory effects [18]. AMPK limits the interleukin 6 (IL-6)-mediated inflammatory response by inhibiting STAT3 activation [19]. STAT3 signaling is required not only for Th17 cell differentiation, but also for T follicular helper (Tfh), memory B, and plasma cell differentiation and antigen-specific antibody responses, suggesting that AMPK activators can also target B cell functions [20–23]. Furthermore, AMPK-dependent inhibition of STAT3 and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κBs) pathways are involved in the inhibition of monocyte-to-macrophage differentiation, and reduce the production of inflammatory cytokines by macrophages [24,25]. Therefore, AMPK activation could be a therapeutic option in autoimmune and inflammatory disorders by modulating multiple intracellular signaling pathways. In this review, we discuss the mechanistic role of metformin in the treatment of rheumatoid arthritis (RA), osteoarthritis (OA), and other autoimmune inflammatory rheumatic diseases.
THERAPEUTIC EFFECTS OF METFORMIN IN RHEUMATOID ARTHRITIS
RA is a chronic autoimmune disease characterized by synovial hyperplasia and inflammation (synovitis), as well as cartilage and bone destruction in the joints. The interaction of environmental and genetic factors triggers the modification of peptides and failure of immune tolerance, leading to the activation of T and B cells and production of autoantibodies [26]. These activated cells and autoantibodies migrate into the joints and cause an immune response that leads to localized joint inflammation. Innate and adaptive immune cells, cytokines, chemokines, and fibroblast like synoviocytes (FLSs) are involved in the development and perpetuation of synovitis. In addition, FLSs and osteoclasts mediate cartilage and bone destruction, respectively. Therefore, inhibiting these cells in RA patients may have therapeutic benefits.
There is increasing evidence implicating T cells in the pathogenesis of RA. T cells (mostly CD4+ T cells) are abundant in the RA synovium and have intimate contact with other cells [27]. As T cells help B cells to produce autoantibodies, participate in proinflammatory cytokine production, and induce osteoclast activation and bone erosion in RA [28], T cell inhibition might have great benefits in RA treatment. Abatacept (cytotoxic T lymphocyte associated protein 4 [CTLA4]-immunoglobulin), which inhibits the costimulatory signals required for T cell activation, has proven efficacy in RA. However, inhibition of T cells by cyclosporine or anti-CD4 antibodies demonstrated limited efficacy in RA [29]. This suggests that differential targeting of CD4+ T cell subtypes, focusing on the deletion of effector T cells, not Treg cells, may be required for the treatment of RA. Th17 cells are a CD4+ T cell subset that play a role in RA pathogenesis by stimulating local inflammation and mediating cartilage and bone destruction [30,31]. However, Treg cells exhibit defective function in RA, which is restored by anti-tumor necrosis factor (TNF) treatment [32–34]. These results suggest that an imbalance between Th17 and Treg cells is a hallmark of RA, and maintaining the balance between these cells would reduce disease severity. Modulation of the Th17-Treg cell imbalance is possible by suppressing cytokines, certain cells, or intracellular signaling pathways. Th17-Treg cell imbalance in RA can be reversed by anti-TNF [33–36] and anti-IL-6 receptor [37,38] treatment. STAT3 inhibition using siRNA reduced Th17 cell differentiation and enhanced Treg cell differentiation among CD4+ T cells isolated from the synovial fluids and peripheral blood of RA patients [39]. Therefore, metformin, a STAT3 inhibitor, may exert therapeutic effects in RA by regulating the Th17-Treg cell imbalance. Kang et al. [40] first reported that metformin dose-dependently reduced arthritis severity in mice with collagen-antibody-induced arthritis. Metformin delivered by intraperitoneal injection significantly reduced Th17 cell differentiation, where this effect was associated with the downregulation of mTORC1 and STAT3 through the activation of AMPK. Autoimmune arthritis also improved with oral administration of metformin in mice with collagen-induced arthritis (CIA) [41]. Th17 cell differentiation was decreased and Treg cell differentiation was enhanced by metformin treatment in CIA mice and in vitro experiments.
Synovial pannus is a fibrovascular tissue with a tumor-like, aggressive phenotype that facilitates joint destruction in RA [27]. As a major component of the synovial pannus, FLSs plays a central role in pannus-mediated joint destruction. RA FLSs can proliferate in an anchorage-independent manner, escape contact inhibition, resist apoptosis, and migrate and adhere to the cartilage matrix (and subsequently damage it by producing matrix-degrading enzymes) [42]. Therefore, drugs affecting RA FLSs could have therapeutic efficacy in RA. Metformin has been shown to inhibit the proliferation and migration of RA FLSs [43,44]. Metformin-induced cell cycle (G2/M phase) arrest, but not apoptosis, of RA FLSs in vitro [43]. This inhibitory effect on RA FLS proliferation was mediated by phosphoinositide 3-kinase (PI3K)-AKT-mTOR pathway inhibition. Treatment with a PI3K inhibitor also inhibited RA FLS proliferation, whereas the use of a PI3K activator had the opposite effect. These results are in line with the fact that the PI3Kδ isoform, expressed by FLSs, is a regulator of FLS growth, migration, and invasion, and may be a potential target for RA therapy [45,46]. In addition to the inhibitory effects on the proliferation and migration of RA FLSs, metformin also downregulated the production of inflammatory cytokines by RA FLSs [44]. Histologically, metformin treatment inhibited synovial hyperplasia, inflammatory cell infiltration, and neovascularization, and downregulated the expression of matrix-degrading enzymes in CIA rat cartilage, confirming its protective effects against synovitis and articular cartilage degradation [47].
Metformin was protective against bone erosion in RA animal models via inhibition of osteoclast differentiation and activity. Osteoclasts are the major cells responsible for bone destruction in RA. Osteoclast differentiation is stimulated by receptor activator of NF-kB ligand (RANKL), which is produced by activated T cells and FLSs [48,49]. Local proinflammatory cytokines can also mediate osteoclastogenesis by inducing RANKL or directly stimulating osteoclast precursors [50]. Preclinical studies have found that metformin treatment inhibited osteoclast differentiation and bone-resorbing activity, thereby reducing bone erosions in the joints of CIA models [41,47,51,52]. Due to the high energy demands of osteoclast generation and maturation, glycolysis and glutaminolysis are required for osteoclast differentiation and bone resorption [51]. Hypoxia-inducible factor 1α (HIF1α) and c-Myc activation induces glucose and glutamine transporters, and stimulates glycolysis and glutaminolysis, respectively. Furthermore, mTOR is constitutively expressed in osteoclasts, and is required for osteoclastic bone resorption as a nutrient sensor. Treatment with an mTOR inhibitor, Torin1, was found to suppress osteoclast differentiation and bone resorption. Since metformin can suppress mTOR and HIF1α, it could be an inhibitor of osteoclastic bone resorption [41].
Metformin exerts anti-inflammatory effects by regulating autophagy and apoptosis. It was suggested in an earlier study that metformin could exert anti-inflammatory effects by targeting lysosomal proteolysis, similar to chloroquine [53]. The difference was that chloroquine swells lysosomal vesicles and raises the vacuolar pH, whereas metformin binds to metal ions and synergizes their inhibitory action. In another study, metformin improved impaired autophagy to exert anti-inflammatory effects in autoimmune arthritis [54]. Autophagy was observed in a K/BxN serum transfer mouse model, but autophagic flux was markedly impaired. Enhanced autophagic flux by metformin resulted in selective degradation of IκB kinase (a kinase that degrades NF-κB-inhibiting IκB proteins), thereby suppressing NF-κB signaling and subsequent inflammatory responses. In addition, metformin improved mitochondrial dysfunction in mouse models of RA. A combination of metformin and coenzyme Q10 treatment protected mitochondria against H2O2-induced apoptosis, by increasing the mitochondrial membrane potential and mitochondrial respiration compared to metformin or coenzyme Q10 alone [55]. Metformin also recovered mitochondrial dysfunction exacerbated by rapamycin treatment [56].
Metformin improved metabolic profiles in in vivo and ex vivo RA models. As metabolic disorders are known to be associated with a poor treatment response in RA [57,58], improving metabolic dysfunction might also ameliorate autoimmune arthritis. Kim et al. [59] found that metformin normalized metabolic dysfunction in obese CIA mice by inducing fibroblast growth factor 21 (FGF21) expression and brown adipose tissue differentiation. Splenocytes co-cultured with brown adipose tissue reduced Th17 cell differentiation, supporting the therapeutic role of metformin in autoimmune arthritis. The relationship between metabolic disorders and RA disease activity was also determined in an Irish RA cohort [60]. Insulin resistance was associated with synovitis in RA patients [60]. The modulation of glucose metabolism in RA FLSs by metformin reduced production of the inflammatory mediators IL-6, IL-8, and monocyte chemoattractant protein 1 (MCP-1) [60], indicating a role of altered glucose metabolism in the suppression of joint inflammation. Moreover, metformin exerted additional anti-inflammatory effects in animal models of RA when concurrently administered with other drugs, such as coenzyme Q10, omega-3, and LMT-28 (an inhibitor of IL-6 signaling) [55,61,62].
Beneficial effects of metformin on RA were demonstrated in clinical studies of RA patients. Using the National Health Insurance Database from Taiwan, patients with RA and type 2 diabetes who received both cyclooxygenase-2 (COX-2) inhibitors and metformin showed lower admission rates during a 10-year follow-up compared to those treated with COX-2 inhibitors alone [63]. Metformin also affected the onset and outcomes of RA. Adherence to metformin treatment was associated with a reduced risk of RA during an 18-year follow-up in a nationwide retrospective cohort study [64]. In a randomized, double-blind, placebo-controlled clinical trial, patients with active RA were treated with additional metformin (1,000 mg/day) or placebo on background methotrexate therapy [65]. After 12 weeks of treatment, metformin-treated patients showed better clinical outcomes than placebo-treated patients, reflected in significant improvement in disease activity and higher rates of remission [65]. These results indicate that metformin could be a beneficial adjunctive therapy in RA. The preclinical and clinical studies regarding the efficacy of metformin in RA are summarized in Table 1.

Studies regarding metformin efficacy in the treatment of RA
THERAPEUTIC EFFECTS OF METFORMIN IN OSTEOARTHRITIS
OA is a heterogeneous, multifactorial joint disease characterized by cartilage degradation, bone abnormalities (such as osteophyte formation and subchondral bone sclerosis) and low-grade synovial inflammation [66,67]. Multiple risk factors, including aging, mechanical stress, trauma, hormonal changes, metabolic disturbances, and genetic predisposition are involved in the pathogenesis of OA. Obesity is an established risk factor for knee, hip, and hand OA [68], and metabolic syndrome and type 2 diabetes are associated with OA [69,70].
Currently, there are no disease-modifying treatments available for OA. As chronic hyperglycemia and insulin resistance induce oxidative stress, chronic low-grade inflammation, and subsequent damage to cartilage matrix [70], metformin might have therapeutic benefits in patients with metabolic OA. In a cohort study using a UK electronic health record database, metformin did not reduce the risk of OA in patients with type 2 diabetes, after adjusting for age and sex [71]. On the other hand, in a case-control study using the Taiwan National Health Insurance Research Database, patients with OA and type 2 diabetes treated with COX-2 inhibitors and metformin were at lower risk of joint replacement surgery compared to those treated with COX-2 inhibitors alone [72]. Considering a previous study showing that type 2 diabetes was an independent predictor of severe OA requiring joint replacement [73], metformin treatment might reduce structural damage and lead to better clinical outcomes in OA. This was confirmed in a prospective cohort study performed using Osteoarthritis Initiative (OAI) data, which found that metformin significantly attenuated knee cartilage volume loss (as assessed by magnetic resonance imaging) over a 4-year follow-up in obese OA patients [74]. The mechanisms of action of metformin in OA treatment were further investigated in preclinical studies.
In OA, cartilage degradation is primarily mediated by catabolic factors produced by chondrocytes in response to the proinflammatory cytokines IL-1β and TNF (secreted by synovial lining cells or chondrocytes themselves). Major catabolic factors include matrix metalloproteinases (MMPs) and a disintegrin and metalloproteinase with thrombospondin motifs (ADAMTSs); their expression levels are increased in OA cartilage and synovium [75–78]. Given that both MMPs and ADAMTSs were highly expressed in the lesions of OA cartilage, they might play a major role in cartilage matrix degradation in OA. For instance, MMP-13, the most potent protease for type II collagen, was highly expressed in human OA chondrocytes [79], and MMP-13-overexpressing transgenic mice exhibited articular cartilage loss and an OA phenotype [80]. ADAMTS-4 and ADAMTS-5 are the major aggrecanases in human and mouse OA, respectively [81,82], and the deletion of ADAMTS5, and dual deletion of ADAMTS4 and ADAMTS5, prevented cartilage degradation in a surgically induced OA mouse model [83,84]. These results indicate that MMP and ADAMTS could be therapeutic targets for the modification of OA.
Metformin reduced the expression of catabolic factors in OA chondrocytes and protected against cartilage degradation in OA animal models. Metformin-treated human OA chondrocytes showed reduced gene expression of the catabolic markers ADAMTS5 and MMP-1 compared to untreated control chondrocytes [85]. The chondroprotective effects of metformin have been demonstrated in an OA mouse model. Articular cartilage degradation developed after surgically induced destabilization of the medial meniscus (DMM) in mice, identified on the basis of higher Osteoarthritis Research Society International (OARSI) scores and smaller cartilage areas [86–88]. Cartilage degradation was significantly reduced in mice administered intragastric or intraarticular metformin [86–88]. Metformin reduced cartilage matrix degradation and stimulated matrix synthesis, as evidenced by a dose-dependent decrease in MMP-13 expression and dose-dependent increase in type II collagen expression in cartilage [86,87]. In another study, mouse articular chondrocytes were treated with metformin or vehicle under IL-1β or TNF-α stimulation in vitro, and metformin-treated chondrocytes demonstrated reduced gene expression of MMP-13, MMP-3, ADAMTS4, and ADAMTS5, and increased gene expression of Col2a1 and aggrecan [88]. This inhibition of cartilage catabolism was mediated by AMPK activation. Metformin increased AMPK expression and phosphorylation in murine OA cartilage and chondrocytes [86–88]. The effect of metformin on AMPK activation and MMP-13 inhibition was diminished after an AMPK inhibitor was added to metformin-treated IL-1β-stimulated mouse chondrocytes [86], and the chondroprotective effect of metformin disappeared in AMPKα1 knockout mice [88]. Given that the deletion of AMPK increased the severity of surgically induced OA in mice [89], these results suggest that maintaining AMPK signaling might be important for preventing OA development and progression, and that metformin could be a potential therapy in OA.
Metformin activated autophagy and inhibited the apoptosis of chondrocytes to attenuate cartilage degradation. There is a direct relation between chondrocyte apoptosis and cartilage degradation [90]. A reduced number of cells and reduced ability to synthesize qualified cartilage matrix contribute to OA initiation and progression. Chondrocyte apoptosis may be induced by mechanical injury, oxidative stress, impaired mitochondrial function, reduced autophagy, and impaired regulation of cell senescence [91,92]. Autophagy inhibition caused OA-like changes in gene expression of chondrocytes, whereas its induction prevented OA-like changes by reducing oxidative stress and apoptosis [93]. Wang et al. [94] demonstrated that metformin reduced oxidative stress and enhanced mitochondrial function in IL-1β-stimulated mouse chondrocytes by promoting phosphatase and tensin homolog (PTEN)-induced putative kinase protein 1 (PINK1)/Parkin-dependent mitophagy. Metformin enhanced PINK1/Parkin-dependent mitophagy, an autophagic elimination of damaged mitochondria, via upregulation of sirtuin 3 (SIRT3), a mitochondrial deacetylase. Activation of SIRT3-mediated PINK1/Parkin-dependent mitophagy had protective effects against OA cartilage catabolism. Another action of metformin was activation of AMPK/SIRT1-mediated autophagy in chondrocytes [95]. Metformin attenuated cartilage degradation, and also enhanced autophagy and inhibited chondrocyte apoptosis, in the articular cartilage tissue of DMM-induced OA mice. Silencing AMPKα2, but not AMPKα1, reduced the expression of SIRT1 and abolished metformin-induced autophagy activation, suggesting that the AMPKα2-SIRT1 pathway was involved in the autophagy-augmenting effects of metformin in chondrocytes. The regulation of autophagy and apoptosis by metformin was replicated in monosodium iodoacetate-induced OA mice and in vitro human OA chondrocytes [96]. Metformin also delayed aging of articular chondrocytes, as demonstrated by reduced expression of cell senescence markers [87].
Metformin decreased synovitis and osteophyte formation in OA animal models. Histologic grading and micro-computed tomography analysis demonstrated synovial hyperplasia and osteophyte formation at 6 and 12 weeks after DMM surgery in mice [88]. Metformin markedly reduced the synovitis score, and osteophyte size and maturity [88]. IL-1β and TNF produced by OA chondrocytes stimulate chondrocytes and synovial cells to produce other proinflammatory mediators and catabolic factors, leading to cartilage destruction. Cartilage degradation products in turn amplify synovial inflammation, and the activated synovium produces proteases that damage adjacent cartilage, forming a vicious cycle [97]. Therefore, decreasing synovitis might prevent or delay OA progression. Metformin reduced joint and systemic inflammation by inhibiting NF-κB, which was involved in inflammatory OA cascades [88,98,99]. In addition to reducing synovitis and structural damage, metformin also decreased pain levels in OA animal models [86,88,96]. The inhibitory action of metformin on pain-related markers in dorsal root ganglion and articular chondrocytes was also mediated by AMPK signaling activation [88]. Moreover, concomitant administration of metformin augmented the anti-inflammatory and chondroprotective effects of other treatments, such as mesenchymal stem cells (MSCs) and COX-2 inhibitors, in experimental OA, suggesting clinical applications for OA treatment [96,100]. The preclinical and clinical studies assessing the efficacy of metformin in OA are summarized in Table 2.
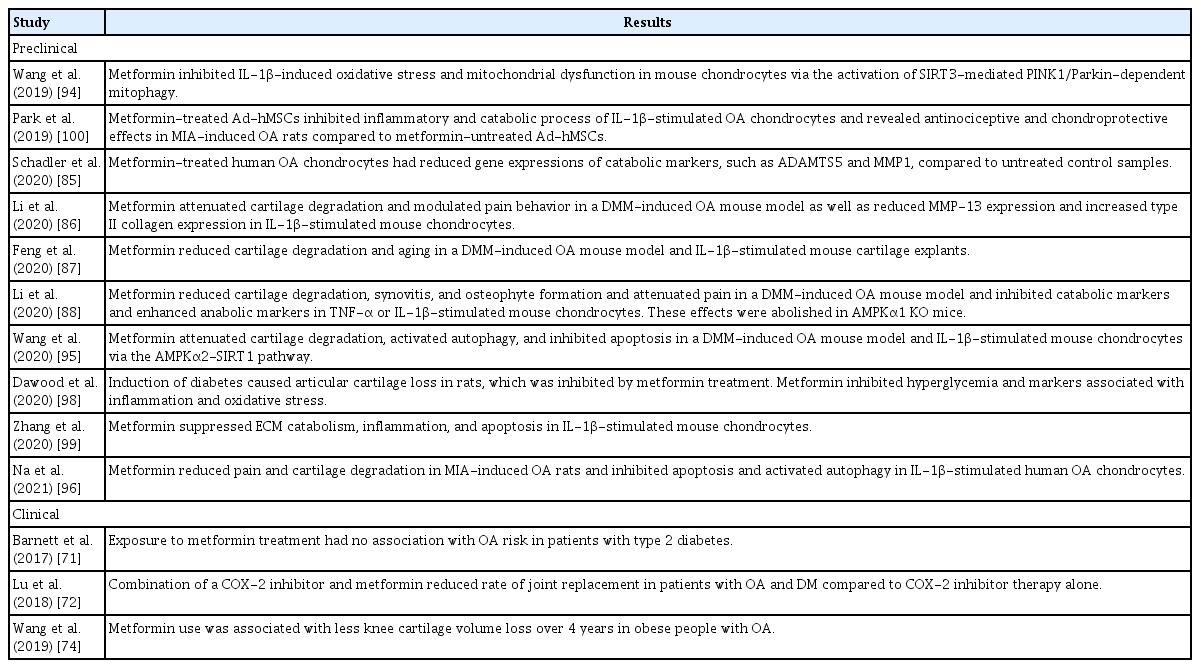
Studies regarding metformin efficacy in the treatment of OA
THERAPEUTIC EFFECTS OF METFORMIN IN OTHER AUTOIMMUNE INFLAMMATORY RHEUMATIC DISEASES
Systemic lupus erythematosus (SLE) is an autoimmune disease that affects multiple organs and is characterized by activation of the type 1 interferon (IFN) pathway, autoreactive T and B cell proliferation, autoantibody production, and immune complex deposition [101,102]. CD4+ T cells from mouse models of and patients with SLE exhibited increased glycolysis and mitochondrial oxidative phosphorylation, reflecting CD4+ T cell activation [103]. Combined treatment with 2-deoxy-D-glucose, a glycolysis inhibitor, and metformin, a mitochondrial oxidative phosphorylation inhibitor, normalized lupus CD4+ T cell effector functions in vitro and reversed the lupus phenotype in mice. A subsequent study found that pyruvate oxidation and oxidative phosphorylation, rather than the pyruvate reduction to lactate, were important for CD4+ T cell activation in lupus-prone mice [104]. The inhibition of pyruvate reduction to lactate, which is an anaerobic process, did not reverse or prevent the lupus phenotype in mice. These results indicate that targeting glucose oxidation can be a novel therapeutic strategy for SLE.
Metformin can inhibit the type 1 IFN pathway. Wang et al. [105] observed enhanced release of mitochondrial DNA (mtDNA) from neutrophil extracellular traps (NETs) and increased levels of antibodies against mtDNA in SLE patients. Correlated with SLE disease activity and lupus nephritis, mtDNA and anti-mtDNA induced IFN-α from plasmacytoid dendritic cells (pDCs) to a significantly greater degree compared to any other DNA antigen or autoantibody. Metformin treatment downregulated NET mtDNA release from neutrophils and decreased IFN-α production from pDCs [105]. Metformin also inhibited the type 1 IFN response in CD4+ T cells in healthy individuals and SLE patients, by suppressing STAT1 phosphorylation and its downstream signaling [106]. These effects of metformin relied on an inhibitory action against mitochondrial oxidative phosphorylation and were independent of AMPK and mTORC1 signaling.
Metformin controlled adaptive immune responses in SLE animal models. In experiments with murine SLE, metformin decreased autoantibody titers and target organ inflammation, including lupus nephritis [107]. Metformin inhibited germinal center reaction, controlled B cell differentiation into plasma cells, and regulated T cell differentiation by reducing Tfh and Th17 cells and increasing Treg cells. The regulation of B and T cells by metformin was associated with AMPK activation, and mTOR and STAT3 inhibition. In addition, metformin enhanced the immunomodulatory effects of other treatments. A recent study by Jang et al. [108] demonstrated enhanced immunoregulatory actions of metformin-treated MSCs compared to metformin-untreated MSCs. Metformin upregulated STAT1 expression to enhance the immunoregulatory actions of MSCs; these effects were mediated by the AMPK and mTOR pathways. The administration of metformin-treated MSCs improved dysregulated B and T cell responses and the lupus phenotype in mice [108]. Another study investigated the therapeutic effect of metformin in combination with abatacept in murine SLE [109]. Although abatacept did not have any efficacy in lupus nephritis, the combination of metformin and abatacept, administered early in the disease course, reduced renal pathology and prevented lupus nephritis, as well as suppressing CD4+ T cell effector subsets. When metformin (500 mg three times per day) was added to standard treatment with glucocorticoids and/or immunosuppressive agents in SLE patients in randomized clinical trials, the risk of SLE flares and required dose of glucocorticoids were reduced [105,110].
Therapeutic effects of metformin were also investigated in Sjögren’s syndrome [111], scleroderma [112–115], ankylosing spondylitis [116], and gout [117]. Metformin reversed salivary gland inflammation and hypofunction in murine Sjögren’s syndrome by controlling imbalanced T and B cell differentiation [111]. To examine the effects of metformin on scleroderma, a mouse model of bleomycin-induced skin fibrosis was employed [112–115]. Metformin attenuated skin fibrosis in mice, as evidenced by reductions in skin thickness, collagen deposition, and numbers of myofibroblasts. Metformin-treated mice had increased Treg cells, and decreased Th17 cells, Tfh cells, germinal center B cells, and proinflammatory cytokine production, along with mTOR and STAT3 suppression in skin fibroblasts. As gout is an inflammatory arthritis induced by monosodium urate (MSU) crystals deposited in tissues, including joints, and where the inflammation is mediated by mTOR signaling in monocytes, inhibition of mTOR might have therapeutic benefits in gout [117]. mTOR inhibition by metformin resulted in reduced MSU crystal-induced monocyte death (pyroptosis) and inflammation in vitro; gout patients exhibited a decreased frequency of gout attacks [117].
CONCLUSIONS
The antidiabetic mechanisms of metformin are still being elucidated, despite its widespread, long-term use in type 2 diabetes. As metformin regulates cell metabolism, proliferation, growth, and autophagy, it might have disease-modifying effects in various other conditions. Metformin has shown therapeutic benefits in obesity, aging, cardiovascular diseases, liver diseases, renal diseases, and cancers [118]. In this review, we focused on the therapeutic applications of metformin in autoimmunity and inflammation, and discussed how metformin impacts RA, OA, and other autoimmune inflammatory rheumatic diseases. Potential molecular and cellular mechanisms of action of metformin in RA and OA are summarized in Fig. 1. Metformin inhibits the proliferation and migration of FLS, regulates Th17 and Treg cell differentiation, suppresses osteoclast differentiation and activity, and reduces inflammatory cytokine production. Metformin can also enhance autophagy and improve mitochondrial functions. Metformin decreases chondrocyte apoptosis and cartilage catabolism. These effects are mediated by the modulation of multiple interacting intracellular pathways, such as PI3K-AKT-mTOR, mTOR, STAT3, HIF1α, NF-κB, and SIRT signaling. By controlling synovitis and joint destruction in RA, and attenuating cartilage degradation in OA in vitro and in vivo, metformin has demonstrated therapeutic benefits in both diseases. Metformin enhances the immunomodulatory, anti-inflammatory, and chondroprotective effects of conventional treatments, suggesting its role as an adjunctive therapy. In clinical studies, metformin treatment was associated with a reduced risk and severity of RA and OA. Metformin reduced the rates of admission and joint replacement surgery, and attenuated cartilage damage in obese and diabetic patients with RA or OA. These results suggest that metformin may have clinical efficacy in RA and OA; however, further clinical studies in large populations are warranted to support its use in a clinical setting.

Potential molecular and cellular mechanisms of metformin in rheumatoid arthritis (RA) and osteoarthritis (OA). Metformin inhibits mitochondrial respiratory chain complex 1, which results in depleted energy status, as represented by the increased adenosine monophosphate (AMP)/adenosine triphosphate (ATP) ratio. This causes the activation of AMP-activated protein kinase (AMPK). AMPK modulates cellular metabolism, proliferation, growth, and autophagy through multiple intracellular signaling pathways. Metformin inhibits proliferation and migration of RA fibroblast like synoviocytes (FLSs), regulates Th17 and regulatory T (Treg) cell differentiation, suppresses osteoclast differentiation, and reduces inflammatory cytokine production, thus controlling synovitis and joint destruction in RA. Metformin also enhances autophagy, mitophagy, and mitochondrial function, leading to decreased apoptosis. Inhibitory actions of metformin against chondrocyte apoptosis and cartilage catabolism attenuate cartilage degradation in OA. PI3K, phosphoinositide 3-kinase; mTORC, mammalian target of rapamycin complex; HIF1α, hypoxia-inducible factor 1α; STAT, signal transducer and activator of transcription; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cell; SIRT, sirtuin; MMP, matrix metalloproteinase; ADAMTS, a disintegrin and metalloproteinase with thrombospondin motif.
Acknowledgments
This work was supported by a grant of the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health and Welfare, Republic of Korea (grant number HI20C1496), and was also supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (Ministry of Science and ICT) (No. 2019R1G1A1100421).
Notes
No potential conflict of interest relevant to this article was reported.