A genome wide association study for lung function in the Korean population using an exome array
Article information

Abstract
Background/Aims
Lung function is an objective indicator of diagnosis and prognosis of respiratory diseases. Many common genetic variants have been associated with lung function in multiple ethnic populations. We looked for coding variants associated with forced expiratory volume in 1 second (FEV1) and FEV1/forced vital capacity (FVC) in the Korean general population.
Methods
We carried out exome array analysis and lung function measurements of the FEV1 and FEV1/FVC in 7,524 individuals of the Korean population. We evaluated single variants with minor allele frequency greater than 0.5%. We performed look-ups for candidate coding variants associations in the UK Biobank, SpiroMeta, and CHARGE consortia.
Results
We identified coding variants in the SMIM29 (C6orf1) (p = 1.2 × 10–5) and HMGA1 locus on chromosome 6p21, the GIT2 (p = 6.5 × 10–5) locus on chromosome 12q24, and the ARHGEF40 (p = 9.9 × 10–5) locus on chromosome 14q11 as having a significant association with lung function (FEV1). We also confirmed a previously reported association with lung function and chronic obstructive pulmonary disease in the FAM13A (p = 4.54 × 10–6) locus on chromosome 4q22, in TNXB (p = 1.30 × 10–6) and in AGER (p = 1.09 × 10–8) locus on chromosome 6p21.
Conclusions
Our exome array analysis identified that several protein coding variants were associated with lung function in the Korean population. Common coding variants in SMIM29 (C6orf1), HMGA1, GIT2, FAM13A, TNXB, AGER and low-frequency variant in ARHGEF40 potentially affect lung function, which warrant further study.
INTRODUCTION
Lung function is an important trait of the respiratory system. Lung function measurements of the forced expiratory volume in one second (FEV1) and the ratio of FEV1 to forced vital capacity (FEV1/FVC) are used as criteria for chronic obstructive pulmonary disease (COPD) diagnosis and severity evaluation for pulmonary disease [1,2]. Although environmental factors such as smoking, air pollution and particulate matter influence lung function, the heritability of lung function has been reported to be around 40% [3,4]. Genome wide association studies (GWASs) for lung function have been reported in data on large populations [5,6]. As expected, genetic loci associated with lung function were shown to play roles in susceptibility to respiratory disease including COPD [7]. However, most identified variants through GWASs are common variants (minor allele frequency [MAF] > 5%) of the population. As in many other complex traits, despite the extensive discovery of associated loci from GWAS, there are some limitations in understanding diseases risk or trait variability only through association of common variants [8]. This problem, so called missing heritability, might be explained by low-frequency and rare variants, and structural variation [8,9].
The exome array contains mostly variant that alter nonsynonymous, splice or stop codons that are likely to affect protein structure and function. The majority of variants are low-frequency (1% < MAF ≤ 5%) and rare (MAF < 1%) [8,9], which could explain additional disease risk and trait variability. Genotyping using an exome array can be a cost-effective and efficient strategy compared to whole exome sequencing [8]. GWAS results using exome arrays have been reported in COPD [9,10] and as meta-analysis for lung functions in persons with European ancestry [11]. However, these studies included only a small fraction of the Asian population samples. There was a study for exome chip quality control for variant analysis and several more loci were identified using exome array in Korean samples [12]. To gain further insight into genetic influence on lung function and to discover variants in coding regions associated with lung function in the Korean population, we carried out a GWAS using exome-based genotyping array.
METHODS
Study populations
We investigated an exome array for coding variants associated with lung function measurement in 7,524 individuals from the Korean Genome and Epidemiology Study (KoGES), which consists of six prospective cohort studies [13]. Among them, the Korea Association Resource cohort was a population-based cohort from the Ansung rural area and Ansan city in South Korea (KoGES Ansan and Ansung study) that was initiated in 2001. More than 260 traits were examined by means of epidemiological surveys, physical examinations and laboratory tests including a pulmonary function test [14]. Spirometry was carried out in accordance with American Thoracic Society/European Respiratory Society guidelines [15]. The baseline examinations have been previously described [14]. Written informed consents were provided by all participants in this study. The study was conducted with bioresources from National Biobank of Korea, the Centers for Disease Control and Prevention, Republic of Korea (KBN 2017-003) and approved by the Institutional Review Board of Asan Medical Center (2015-1341).
Genotyping and quality of control
In this study, genomic DNAs isolated from peripheral blood were genotyped on the Infinium Human Exome BeadChip v1 (Illumina, San Diego, CA, USA). Genotyping process and quality control of the genotype dataset were previously reported [12]. After quality control, a total of 48,187 single nucleotide polymorphisms (SNPs) were used in the exome array analysis.
Single variant analysis for association with lung function
Single variant association tests for FEV1 and FEV1/FVC were carried out using the linear mixed model. We used the likelihood ratio test implemented in the Genome wide Efficient Mixed Model Association (GEMMA) software package [16]. The fixed effects of each variant was tested after adjusting for age, sex, ever-smoking, packyears, and height. A p < 10-4 was the criterion for single variant association analysis. Variants analysis and annotation of genes was done with the GRCh37/hg19 database.
Gene-based testing for association with lung function
We carried out gene-based analysis using Sequence Kernel Association tests (SKAT) [17] to assess the joint effect of multiple low-frequency and rare genetic variants within genes on lung function traits. SKAT analyses identified the top 10 candidate genes associated (p < 2.5 × 10–5) with FEV1 and FEV1/FVC.
Replication study
We carried out look-up replication of the selected top nine variants for FEV1 and FEV1/FVC in 410,289 subjects in the UK Biobank study (http://biobankengine.stanford.edu), SpiroMeta and Cohorts for Heart and Aging Research in Genomic Epidemiology (CHARGE) (www.chargeconsortium.com) consortia [6,11,18-20]. A p < 10–4 was the criterion for look-ups.
Characterization of findings
We assessed whether the identified loci contained variants associated with gene expression in various tissues by querying the expression quantitative trait loci (eQTL) database of the Genotype-tissue expression (GTEx project) (https://gtexportal.org/home/) [21]. Potentially deleterious coding variants were identified by Sorting Intolerant From Tolerant (SIFT) and PolyPhen-2 [22]. We searched for evidence of protein expression in the respiratory system by querying the Human Protein Atlas (www.proteinatlas.org) [23].
RESULTS
Cohort characteristics
Our analysis included 7,524 individuals (3,942 females; 52.4%) from KoGES with non-missing covariate and lung function phenotypes. The characteristics of the 7,524 individuals who were assessed for an exome array are shown in Table 1. The mean age was 52.1 years. A 40.5% (n = 3,051/7,524) had ever smoked with mean pack-years of 9.0.

Demographic characteristics of study populations
Single variant analysis for association with lung function
We analyzed the association between SNPs from the KoGES exome array data and lung function measures, FEV1 and FEV1/FVC. For primary discovery analysis, we used genotyping data for the 7,524 subjects in the KoGES. First, we checked for sample quality. We detected 19 pairs of samples with genetic relatedness greater than 0.25, and removed one sample from each related pair. There was no sample failed for the genotype missingness test (missing rate < 5%). Next, we checked for marker quality. We removed 1 SNP which failed to pass 95% genotyping rate threshold, 74 SNPs which failed at Hardy-Weinberg Equilibrium (HWE) test in controls (p < 0.0001), and 3 SNPs which were detected as principal component analysis outliers. After quality controls, 77,397 SNPs remained. Plink version 1.07 was used for quality controls procedures and Genome-wide Complex Trait Analysis (GCTA) version 1.26 was used for calculating genetic relationship matrix. In the GEMMA analysis, only 31,571 SNPs were used which passed the internal filters of GEMMA in the default setting.
Finally, we isolate the 31,571 SNPs for FEV1 and 16,616 SNPs for FEV1/FVC. Among them, 10,513 of 31,571 (33.2%) SNPs for FEV1 were rare and low-frequency variants and the rest 21,058 (66.7%) were common variants. Also, 5,294 of 16,616 (31.8%) SNPs for FEV1/FVC were rare and low-frequency variants and the rest 11,322 (68.2%) SNPs were common variants. The top SNPs for FEV1 and FEV1/FVC ratio identified in the KoGES general population are listed in Table 2. Only one genotyped SNP met the exome-wide significance criteria (p < 5 × 10–8) in our exome array analyses. The strongest signal (p = 1.2 × 10–5) for FEV1 was a variant rs1150781 in small integral membrane protein 29 (SMIM29 [C6orf1]) on chromosome 6 (Figs. 1 and 2). Two SNPs (rs7742369, rs2780226) also on chromosome 6 (6p21) were located in or near SMIM29 (C6orf1) and high-mobility group AT-hook 1 (HMGA1). One of the top nine SNPs, variant rs114591848 was a low-frequency variant in rho guanine nucleotide exchange factor 40 (ARHGEF40) on chromosome 14 (MAF = 1.4%) and the others were common variants (Table 2).

Top nine coding variants associated with FEV1, FEV1/FVC and results of look-up analyses
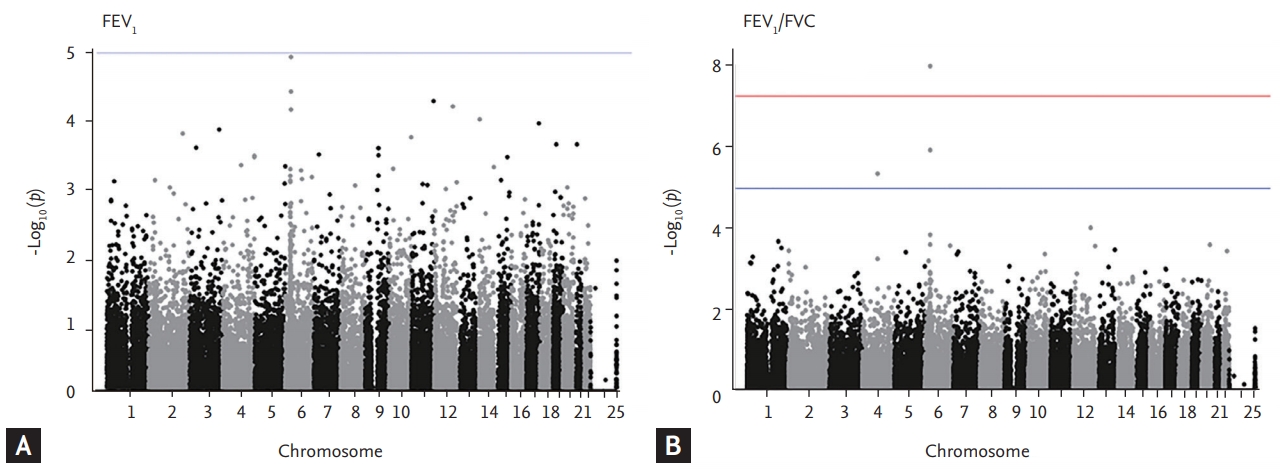
Manhattan plots of association results for forced expiratory volume in 1 second (FEV1) and FEV1/forced vital capacity (FVC). The manhattan plots for (A) FEV1 and (B) FEV1/FVC are ordered by chromosome position. Single nucleotide polymorphisms for which –log10 p > 5 are indicated in blue line.
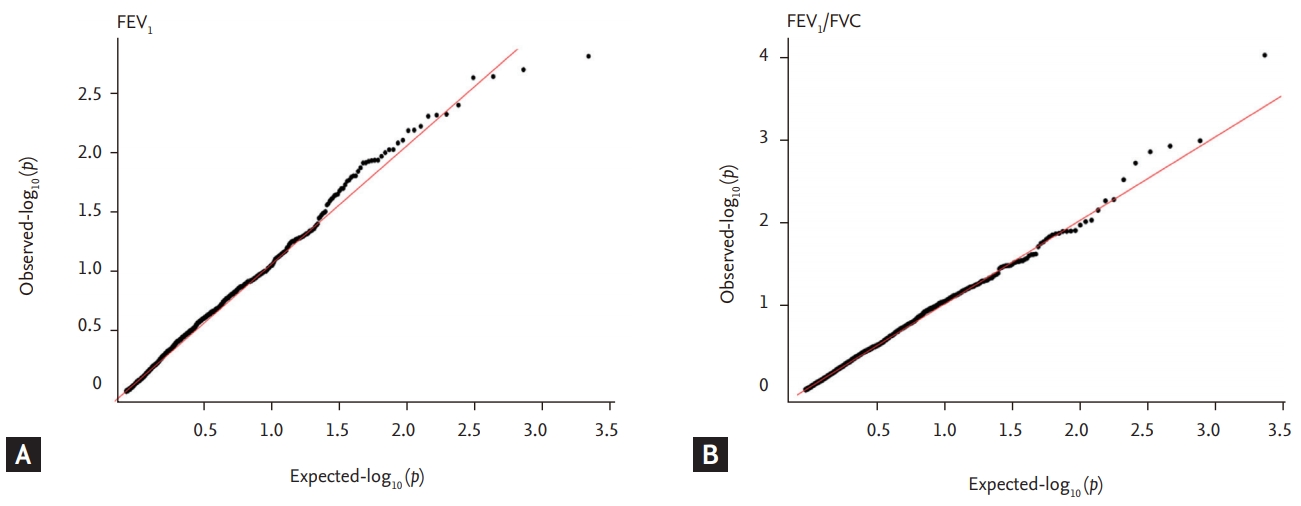
Quantile-Quantile (QQ) plots show –log10 (p) of observed genome wide association results against expected association results for (A) forced expiratory volume in 1 second (FEV1) and (B) FEV1/forced vital capacity (FVC). Genomic control inflation factors (λGC) before genomic control was 0.90 for FEV1 and 0.84 for FEV1/FVC.
The strongest signal (p = 1.0 × 10-8) for FEV1/FVC was a rs2070600 in advanced glycosylation end-product specific receptor (AGER) on chromosome 6, which previously reported as a locus associated with lung function and COPD [18,19]. The second strongest signal (p = 1.3 × 10-6) was rs2239688 in tenascin XB (TNXB) on chromosome 6. TNXB encoded extracellular matrix glycoproteins, which are associated with organizing and maintaining the structure of tissues that support the body’s muscles, joints, organs, and skin. This gene was previously reported to be associated with IPF [24] and COPD, lungfunction [25]. To gain further insight into the associated variants, we assessed whether the candidate variants, or their proxies were associated with gene expression in various tissues by using GTEx and eQTL analyses (Supplementary Table 1). Among them, sentinel variants or their close proxies of rs114591848, rs7671167, and rs2070600 variants were eQTL in lung for ARHGEF40, family with sequence similarity 13 member A (FAM13A), and AGER. The protein and mRNA expression profiles of all implicated genes from the single variant association analyses are shown in Supplementary Table 2.
We also carried out a look-up in the publicly available UK Biobank results as adjusted for sex and ancestral principle components, SpiroMeta, and CHARGE consortia data in order to confirm the novelty of our results and presence or absence of difference of genetic variation in lung function among different ethnicities. SNPs associated with lung function within ± 1.5 Mb regions from selected variants were presented. This look-up showed evidence of replication for variant rs1150781 in or near SMIM29 (C6orf1) and HMGA1 on chromosome 6, proxies of variants rs114591848 in ARHGEF 40 on chromosome 14 for FEV1, and variant rs7671167 in FAM 13A and variant rs2070600 in AGER on chromosome 6 for FEV1/FVC (Table 2).
Gene-based analysis for gene association with lung function
For gene-based analysis, we carried out the SKAT method to assess the joint effects of variants within genes on lung function traits. The top 10 most significant genes and p values of lung function are shown in Table 3. Our top association was in the gene DNA fragmentation factor subunit alpha (DFFA) (p = 8 × 10–8 for FEV1, p = 5.8 × 10–18 for FEV1/FVC). However, we confirmed that the SKAT analysis and the candidate SNPs in or near target genes did not match.

Association results for all genes identified in SKAT analyses (KoGES)
DISCUSSION
In this study, we identified three loci (chromosome 4, 6, and 14) associated with lung function. A look-up study revealed that our novel SNPs in 6p21 and 14q11 loci replicated the association with FEV1 from the UK Biobank. Some of the candidate SNPs (rs7742369, rs2780226, rs1150781, rs2239688, and rs2070600) were located on 6p21. We previously reported that this locus on 6p21 influences lung function in the Korean population [14]. The variant rs1150781 (MAF = 18%, p = 1.2 × 10–5, Gly150Ala, PolyPhen prediction: benign) (Supplementary Table 3) is a missense variant in SMIM29 (C6orf1), which encodes an integral membrane and is expressed in brain, skin, thyroid, spleen, and lungs. This protein consists of 102 amino acids with molecular weight is of 11.5 kDa and is detected in human fetal lung cell lysate and respiratory epithelial cells. The expression of this protein was reported to increase in some non-small cell lung cancer patients, especially for adenocarcinoma and squamous cell lung cancer [23]. However, gain and loss of functional studies of this gene are lacking. For this reason, although our exome array analysis identified the missense variant rs1150781 and nonsynonymous substitution (Gly150Ala) of the SMIM29 (C6orf1) protein, to determine whether variant rs1150781 affects protein function, further validation of the association and functional studies of SMIM29 (C6orf1) will be required. Variant rs1150781 and their proxy (rs2780226, LD, r2 = 0.99) were located in or near SMIM29 (C6orf1) and HMGA1. SMIM29 (C6orf1) was located downstream of HMGA1, and these two genes are related to genetic linkage. HMGA1 encodes a protein related to epigenetic modification and functions as a dynamic regulator of chromatin structure and transcription, which is localized in the cell nucleus. The HMGA1 protein is expressed in human lung macrophages and respiratory epithelial cells [23]. Recently, Zhang et al. [26] reported that the protein and mRNA of HMGA1 were highly expressed in intact human airway epithelia and their basal cells. In a loss of function study with HMGA1 siRNA, they demonstrated that HMGA1 down regulation in human airway basal cells led to increase expression of airway remodeling related genes. The NHGRI-EBI catalog of published GWASs shows that variants in or near HMGA1 are associated with body height, BMI and smoking behavior (Supplementary Table 4) [27]. Also, the HMGA1 protein is a key regulator of the insulin pathway [28] and variants of the HMGA1 gene are associated with type 2 diabetes mellitus [29].
We also identified a nonsynonymous variant rs114591848 in the ARHGEF40 locus on chromosome 14. This variant is a low-frequency (MAF = 1.4%) missense variant and resulted in an amino acid change (Arg1062Gln, PolyPhen prediction: possibly damaging) (Supplementary Table 3). ARHGEF40 encodes Rho guanine nucleotide exchange factor is directly responsible for the activation of Rho-family GTPase, and regulates numerous cellular responses such as proliferation, differentiation, and cytoskeletal organization [30].
Moreover, we detected nominal levels of significance with two intronic SNPs rs7671167 in the FAM13A on chromosome 4q22.1, and rs2239688 in TNXB on chromosome 6p21.3 and one exonic SNP rs2070600 in the AGER on chromosome 6p21.3. These variants were previously reported to be loci associated with lung function and pulmonary diseases [6,10,19,20]. The FAM13A isoform 1 protein has a Rho GTPase-activating protein (GAP) domain and participates in the Rho GTPase signaling pathway [31]. Also, ARGEF40 encodes the Rho guanine nucleotide exchange factor. These results suggest that the Rho GTPase signaling pathway might play a role in lung function and COPD.
By means of our exome array analysis, we have tried to identify the low-frequency and rare variants potentially associated with lung function in order to uncover the missing heritability of lung function. However, our discovery analyses did not identify many rare and low-frequency coding variants that are responsible for the lung function trait in the Korean population, probably because of our small sample size and limited statistical power. Further confirmation of these associations in a large sample is needed.
We additionally investigated the joint effects of low-frequency and rare variant within genes, on lung function traits, by using the SKAT gene-based test. In these analyses, we identified an exome-wide significant signal (p = 8 × 10–8 for FEV1, p = 5.8 × 10–18 for FEV1/ FVC) in DFFA, which is also known to be an inhibitor of caspase-activated DNase. DFFA protein product is the substrate for caspase-3 and triggers DNA fragmentation during apoptosis [32]. However, this gene was not replicated in the UK Biobank data.
Our study has some potential limitations. First, the sample size is relatively small, and lack of statistical power may be a limitation. Second, we did not provide further evidence for the biological role of the SMIM29 (C6orf1), HMGA1 and ARHGEF40 in lung function. Finally, our exome array identified only coding variants, but cannot provide the roles of noncoding variants in lung function. To date, many studies and meta-analyses including SpiroMeta, CHARGE consortia, and UK Biobank studies have reported nearly 100 loci and many variants associated with lung function and COPD [11,20,33,34]. However, these studies have been exclusively carried out among populations whereas Asian ancestry populations participate with relatively smaller sample size. Therefore, there is a need to perform GWAS information from many people with Asian ancestry in order to better understand the genetic architecture of lung function.
In conclusion, we have newly identified a common coding variant in or near SMIM29 (C6orf1), HMGA1, and one missense low-frequency variant in ARHGEF40, that are associated with lung function. Although a large sample size may be required to strengthen our results, we present additional evidence to support the notion that the genetic contribution to lung function includes polygenic architecture with low-frequency and common genetic variants in the Korean population.
KEY MESSAGE
1. We identified novel single nucleotide polymorphisms associated with lung function in the Korean population. There are: Common coding variant rs1150781 in or near SMIM29 (C6orf1), HMGA1 located on 6p21 and low-frequency variant rs114591848 in ARHGEF40 locus on 14q11, which were associated with FEV1.
2. Common coding variant rs2070600 in AGER located on 6p21.3 associated with forced expiratory volume in 1 second/forced vital capacity with exome-wide significant threshold as previously reported in loci associated with lung function and chronic obstructive pulmonary disease.
Notes
No potential conflict of interest relevant to this article was reported.
Acknowledgements
This research was supported by the National Research Foundation of Korea (2017R1A2B4003790).