Role of asymptomatic hyperuricemia in the progression of chronic kidney disease and cardiovascular disease
Article information
Abstract
Previous research has investigated whether hyperuricemia serves as an independent risk factor for cardiovascular and renal diseases. Hyperuricemia is defined as an abnormally high level of uric acid (UA; i.e., serum urate level > 6.8 mg/dL). Hyperuricemia has been considered a complication of chronic kidney disease (CKD). However, it seems to play a pathogenic role in the progression of renal diseases. There has been increasing focus on the link between hyperuricemia and CKD. The results of randomized controlled trials have implied independent associations between hyperuricemia and the progression of cardiovascular and renal morbidities. These associations may be mediated by renin-angiotensin system activation, nitric oxide synthase inhibition, and macrovascular/microvascular disease development. There remains controversy regarding the use of serum UA level as an indirect index of renal vascular disease. This literature review focuses on the role of asymptomatic hyperuricemia in the progression of CKD, as well as the association between hyperuricemia and cardiovascular disease. It also provides a general overview of the physiological metabolism of UA.
INTRODUCTION
There is ongoing controversy regarding the association between hyperuricemia and cardiovascular disease (CVD). In the year 2020, CVD is considered one of the most common non-communicable diseases and the major cause of mortality in most developing countries [1]. Uric acid (UA) was discovered in the early 1700s during an investigation of a bladder stone. Serum UA is generally elevated in patients with chronic kidney disease (CKD) but has not received substantial attention in such patients. However, in recent years, the results of many studies have implied that hyperuricemia may be independently associated with the progression of CKD. A 5-year follow-up study of 7,078 individuals in Japan found that baseline levels of UA were independent risk factors for CKD after adjusting for age, sex, body mass index, blood pressure (BP), and plasma glucose level [2]. CKD is an important global public health problem. Without early intervention, it progresses to end-stage renal disease. Therefore, factors that aggravate the deterioration of renal function must be identified. The relationship between hyperuricemia and CVD has been established since the early 1900s. In patients with high BP and CVD, high serum UA is a common finding. Furthermore, UA as a cardiovascular risk factor has been addressed in numerous randomized trials and prospective cohort studies [3]. In recent decades, there has been a reappraisal of the relationship between elevated serum UA levels and an increased risk of cardiovascular and renal injuries [4,5].
The prevalence of hyperuricemia has increased significantly with the increasing incidence of CKD. Moreover, the incidence of hyperuricemia has gradually increased in younger individuals and is closely associated with a westernized lifestyle and environment as well as an unhealthy diet [6–8]. Hyperuricemia typically accompanies metabolic syndrome, hypertension (HTN), and CKD. Serum UA levels are considerably dependent on dietary habits, lifestyle, sex, and the use of medications and diuretics [9]. Previous epidemiologic studies suggested that earlier stages of CKD were linearly related to all-cause and cardiovascular mortality [10].
The physiological solubility of UA occurs at approximately 6.4 mg/dL. Before reaching the point of supersaturation, UA-binding proteins act to increase solubility to near 7.0 mg/dL. At this point, serum UA can crystallize within the human body, leading to the development of hyperuricemia [11]. Possible mechanisms by which high serum UA may exacerbate renal injury include oxidative stress, an inflammatory reaction, renin-angiotensin system activation, nitric oxide synthase inhibition, and epithelial-to-mesenchymal transition [12,13]. These changes may induce renal vascular lesions and tubulointerstitial injury. Specific renal pathological changes include renal arteriosclerosis, glomerular HTN, glomerulosclerosis, interstitial lesions, and acute renal injury. Notably, kidney damage caused by hyperuricemia is often associated with HTN and insulin resistance (IR) [14,15].
Febuxostat is a nonpurine xanthine oxidase (XO) inhibitor and a UA-lowering agent widely used in the control and management of hyperuricemia. Multiple studies have shown that febuxostat is not inferior at reducing serum UA, compared to allopurinol (the most commonly used serum UA-lowering agent) [16]. In the Cardiovascular Safety of Febuxostat and Allopurinol in Patients with Gout and Cardiovascular Morbidities (CARES) trial, which enrolled more than 6,000 patients to evaluate the cardiovascular safety of febuxostat compared with allopurinol [17], the results showed no differences between the two drugs in terms of cardiovascular events. However, a slightly elevated risk of cardiovascular mortality was present in the febuxostat group. Recent clinical studies have shown that early management of hyperuricemia in patients with CKD facilitates the regulation of BP and postponement of renal function decline [6].
Febuxostat was first discovered by scientists at the Japanese pharmaceutical company Teijin (Tokyo, Japan) in 1998. In November 2017, the Food and Drug Administration released a warning alert regarding febuxostat and cardiovascular safety, due to an increased risk of cardiac-related death associated with the use of febuxostat [18]. Another warning by the Food and Drug Administration was released in 2019 to discourage the use of febuxostat as a UA-lowering agent [19].
URIC ACID METABOLISM
Because hyperuricemia may act as a CVD risk factor, there is a need to clarify the metabolic process and mechanisms underlying serum UA production. An understanding of the fundamental processes that contribute to the production of UA may provide insight regarding the association between UA and CVD [11]. Notably, UA is the final product of purine nucleotide metabolism. A disturbance in purine metabolism or abnormal renal excretion of UA can affect serum UA level. According to Alegria-Diaz et al. [20], the normal range of plasma UA in men is 237.9 to 356.9 nmol/L, equivalent to 4 to 6 mg/dL. In women, this range is 178.4 to 297.4 nmol/L, equivalent to 3 to 5 mg/dL. Importantly, UA has antioxidant properties, enhances immunity, and maintains BP [21]. Therefore, serum UA levels are much higher in humans than in other mammals. However, excessive UA levels may induce gout, HTN, and other diseases. Thus, UA plays contradictory roles in the body. Uricase is an enzyme that helps to degrade UA into allantoin, which leads UA to aggregate with adenine and guanine (purine components of DNA and RNA). Furthermore, UA is the product of xanthine metabolism via xanthine oxidoreductase activity. Xanthine dehydrogenase and XO are two inter-convertible forms of xanthine oxidoreductase [22].
Xanthine can be degraded into UA and superoxide anion by XO, which may lead to elevated levels of intracellular superoxide anions and subsequent cell envelope damage, as well as enhanced mutagenesis [23]. Xanthine dehydrogenase is the most common form of xanthine oxidoreductase in vivo and generates the reduced form of nicotinamide adenine dinucleotide. However, xanthine oxidoreductase can be converted to XO when it is in a hypoxic environment (Fig. 1) [24]. Moreover, the excessive production of initial reactive oxygen species caused by various stimuli may activate XO, which remains the main source of reactive oxygen species. An experimental study showed that the initial production of mitochondrial reactive oxygen species activated XO in cultured human primary glomerular endothelial cells exposed to a high-glucose environment. Compared with allopurinol, XO is the major source of reactive oxygen species and causes both endothelial dysfunction and activation [25]. Therefore, UA has a dual antioxidant/oxidant role, depending on its environment. The process of UA production in patients can thus promote the progression of CKD.
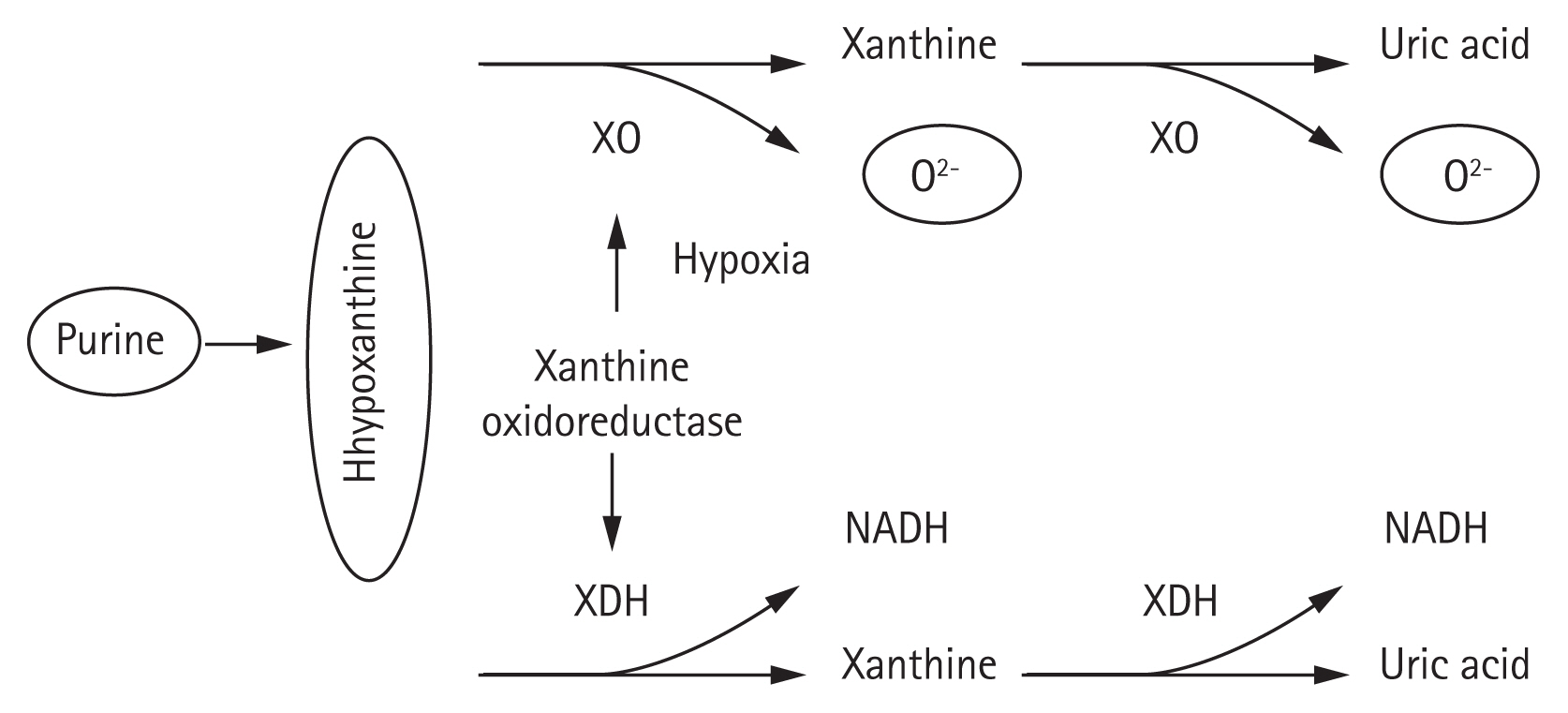
Uric acid metabolism. XO, xanthine oxidase; XDH, xanthine dehydrogenase; NADH, nicotinamice-adenin dinucleotide.
Most energy in the human body is stored in the adenosine triphosphate structure. This structure centers on adenine, which passes through multiple conversion processes to produce hypoxanthine, xanthine, and then UA through the activation of XO and the production of reactive oxygen species during metabolism. These reactive oxygen species tend to bind with nitric oxide, a vasodilator substance, and inhibit its function. This is considered a key factor in the development of serious cardiovascular injury (i.e., arteriosclerosis). During the metabolism of fructose, a large amount of adenosine triphosphate is consumed, resulting in elevated amounts of UA. Recent increases in the rates of HTN and CVD are primarily caused by the elevated intake of sugars including fructose [26]. Ross et al. [27] found that in pathological conditions such as heart failure, oxygen shortage tends to increase the levels of serum lactic acid, which leads to aggravated anaerobic metabolism in tissues. This elevates the lactic acid level and enhances the reabsorption of UA in the kidney, which subsequently increases the serum UA level [27]. Serum UA levels are also influenced by excretion from the kidney and digestive tract. Thus, an increased serum UA level due to the aggravated production of UA could be a marker of systemic circulatory failure. Medications such as XO inhibitors (i.e., allopurinol and febuxostat) are effective for the treatment of hyperuricemia due to aggravated UA production [11].
Research regarding urate transporters
Approximately 60% of UA is biosynthesized in the body and metabolized through the kidney. Therefore, the kidney is the main organ involved in UA excretion. According to Maiuolo et al. [28], abnormal secretion or reabsorption of UA is presumed to contribute to hyperuricemia. Regulation of serum UA levels occurs mainly through glomerular filtration, renal tubule secretion, and reabsorption [28]. Approximately 90% of the UA filtered by the kidney is reabsorbed by proximal tubules, through a process regulated by specific transporters. Several transporters are involved in the renal proximal tubule transport of UA, including glucose transport protein 9 (GLUT9), organic anion transporter, and urate anion transporter 1 (URAT1) [29]. In 2002, Enomoto et al. [30] first discovered the SLC22A12 gene encoding URAT1, a new member of the organic anion transporter family. This protein exhibits unique substrate specificity, compared with other organic anion transporters [30]. URAT1 is an anion-exchanging uptake transporter localized in the apical membrane of renal proximal tubular cells [30,31]. It mediates the reabsorption of UA from the proximal tubule, thereby playing a key role in UA homeostasis. The proximal tubules are responsible for reabsorbing 85% of the filtered UA, and UA transport thus plays an important role in the metabolism of UA [30,32,33]. URAT1 is only expressed at the apical brush edge of renal tubular epithelial cells. The driving force for the transport of UA by URAT1 is the exchange of anions in the cell with UA in the lumen. The accumulated anions in the cells are exchanged with UA in the lumen through the affinity with URAT1. This electrochemical gradient enables UA to complete the reabsorption process. Anions that enter the lumen then pass through glomerular filtration via the lumen membrane and enter the perivascular capillaries by means of organic anion transporters in the basolateral membrane, thus completing cellular metabolism [34,35]. Therefore, a drug with an affinity for URAT1 can promote the excretion of UA via the lumen. By contrast, when such a drug acts on cells in the endothelium, UA reabsorption is promoted and UA excretion is inhibited, thereby regulating the serum UA level. Unlike other ion transporters, URAT1 exhibits specific substrate selectivity and has thus become a new target for regulating the reabsorption of UA. This may offer a new approach for future treatment of hyperuricemia.
Circulation of UA is necessary to protect against oxidative damage, whereas excess serum UA can cause CVD. UA handling by the kidney is complex. Briefly, UA is first eliminated into the proximal tubule by glomerular filtration and/or active uptake via organic anion transporter 1 and elimination from proximal cells by urate channels (e.g., SLC17A3). It is then reabsorbed from the proximal tubule into the cells by URAT1 and (possibly) organic anion transporter 4. Reabsorbed UA is returned to the blood by means of another transporter, GLUT9, or SLC2A9. The accumulated anions in the cells are exchanged with UA in the lumen by means of URAT1 affinity. This electrochemical gradient enables UA to complete the reabsorption process [34,35].
Several factors are involved in the secretion and production of UA. Medications have a considerable impact on changes in UA levels, thus complicating efforts to identify whether fluctuations in serum UA levels are due to UA overproduction or underexcretion. UA and other creatinine clearance tests can be used to determine the cause of underlying disease.
EFFECTS OF UA ON CKD INCIDENCE AND RISK FACTORS
Elevated levels of UA may cause gout, HTN, CVD, and kidney disease, whereas reduced levels of UA are associated with neurodegenerative diseases (e.g., multiple sclerosis, Parkinson’s disease, and optic neuritis) [22]. When UA levels are reduced, the ability to clear a large amount of oxygen free radicals generated by exercise is also reduced, which can damage renal function. Traditionally, renal diseases caused by hyperuricemia are presumed to include acute and chronic UA nephropathy and kidney stones. However, there is considerable evidence that hyperuricemia itself may be associated with the progression of CKD and risk factors for CKD (e.g., HTN and IR) [36].
Hyperuricemia and HTN
In recent years, there has been an increasing number of studies supporting the close relationship between hyperuricemia and cardiovascular events (e.g., HTN). A prospective cohort study by Sundstrom et al. [37] found that hyperuricemia increased the risk of UA one-fold, the risk of HTN 1.17-fold, and the risk of BP progression 1.11-fold. Mellen’s > 9-year follow-up study also showed that serum UA level was positively correlated with HTN [38]. The presumed mechanism was renin-angiotensin system activation, inflammatory reaction, oxidative stress, vascular smooth muscle cell proliferation, and IR.
Activation of the renin-angiotensin system by UA
An animal model study by Mazzali et al. [39] suggested that hyperuricemia-induced activation of the renin-angiotensin system is closely related to the pathogenesis of HTN. Recent clinical studies by Doehner et al. [40]. have also shown that hyperuricemia ultimately leads to HTN by activating the renin-angiotensin system. The uric acid and insulin sensitivity and risk of incident hypertension (ONATA) study [41] indicated that serum UA levels were negatively correlated with insulin sensitivity. Hyperuricemia is often accompanied by IR, which also causes hyperactivity of the renin-angiotensin-aldosterone system and sympathetic nervous system, eventually leading to sodium retention, increased blood volume, and high BP.
Inflammation and oxidative stress induction by UA
Hyperuricemia is accompanied by an elevated level of serum high-sensitivity C-reactive protein [42], which is an important marker of inflammation. This association with an elevated level of serum high-sensitivity C-reactive protein strongly suggests that hyperuricemia is closely related to inflammation and oxidative stress. Soluble urate can participate in vascular inflammation and oxidative stress reactions by increasing low-density lipoprotein oxidation and lipid peroxidation, as well as the high-sensitivity C-reactive protein level. Notably, high-sensitivity C-reactive protein can mediate vascular endothelial necrosis by activating the complement system, causing macrophages and other inflammatory cells to enter the vascular endothelium. This leads to vascular endothelial injury and ultimately promotes the onset and progression of HTN [43].
Effect of UA on the biological behavior of vascular smooth muscle cells
Price et al. [44] showed that although vascular smooth muscle cells do not express UA receptors, they express URAT1, which can facilitate the uptake of UA. Urate can enter into smooth muscle cells through a UA transport channel protein; it then activates specific mitogen-activated protein kinase and induces cyclooxygenase 2 expression [45]. These proteins stimulate the formation of local thrombus and upregulate the expression of platelet-derived growth factor, which enhances vascular smooth muscle cell proliferation and migration [46]. The influence of hyperuricemia on vascular smooth muscle cell biological behavior can lead to vascular remodeling, which contributes to the onset, progression, and resolution of HTN.
Hyperuricemia and IR
IR is the main cause of type 2 diabetes, as well as an important risk factor for HTN, dyslipidemia, and coronary heart disease [47]. Hyperuricemia may also be involved in the development of IR. A study of 7,483 patients without diabetes showed that, following adjustments for the levels of hyperlipidemia, HTN, obesity, and blood sugar, serum UA levels were positively correlated with hyperinsulinemia and the IR index. IR rat model experiments showed that IR rats exhibited increased reabsorption of urate via the upregulation of URAT1 gene expression in the renal cortex. In these rats, the secretion of urate was also reduced through the downregulation of the expression of the urate transporter gene. Treatment with rosiglitazone was able to improve both effects on urate metabolism [48]. A possible mechanism is that elevated serum UA directly damages pancreatic β-cells, resulting in reduced insulin synthesis, diminished receptor sensitivity, and IR. These changes lead to enhanced insulin compensatory secretion. Insulin stimulates the synthesis and secretion of endothelin-1 in aortic endothelial cells [49]. Endothelin-1 is the most potent vasoconstrictor currently known; it can enhance peripheral vascular resistance, promote renal tubular reabsorption of water and sodium, and promote smooth muscle and myocardium proliferation, thus causing cardiovascular remodeling and elevated BP.
Hyperuricemia and CKD progression
Many epidemiologic studies have suggested a role for hyperuricemia in the increased rates of mortality and renal disease, but published data regarding the effects of hyperuricemia on CKD are limited. Hyperuricemia is prevalent in patients with CKD, which may lead to reduced UA excretion during renal functional decline. In recent years, CKD has become a global public health problem because of its high prevalence and the accompanying increased risk of end-stage renal disease. Emerging evidence suggests a pathogenic role for hyperuricemia in the development and progression of CKD [50]. Notably, untreated hyperuricemia is considered a risk factor for the onset of CKD [51,52].
The prevalence of hyperuricemia in patients with CKD in China is 36.6% to 50% and is significantly higher with progression of CKD [7,53]. Patients with stage 4 or 5 CKD require the initiation of dialysis and exhibit a gradual increase in the risk of death. Recent studies have shown that hyperuricemia is actively involved in renal dysfunction [54]. In a 25-year follow-up study of 177,570 patients with nephropathy in the United States, Chonchol et al. [52] found that the risk of CKD in patients with the highest UA level was 2.14-fold greater than the risk in non-CKD patients with normal UA levels. The mechanism by which hyperuricemia contributes to CKD involves renal inflammation, endothelial dysfunction, and activation of the renin-angiotensin system [55]. Moreover, several epidemiological studies have linked hyperuricemia with an increased risk of CKD. Briefly, hyperuricemia is presumed to stimulate the renin-angiotensin system and inhibit the release of endothelial nitric oxide, which leads to renal vasoconstriction and elevated BP. Concurrently, a high UA level plays a pathogenetic role in the onset of inflammation and contributes to the progression of renal disease [56,57].
Urate deposition in renal tubules and renal interstitium can reduce the expression of nitric oxide synthase in the kidney. Nitric oxide plays an important role in regulating the relaxation activity of vascular endothelial cells, maintaining consistent renal vascular tension, and modulating renal tissue blood flow, renin secretion, and tubuloglomerular feedback [58]. Nitric oxide synthase is inhibited by hyperuricemia. In particular, endothelial cells can upregulate angiotensin-converting enzyme activity [59], thus increasing the production of angiotensin II and superoxide anions, which triggers vasoconstriction and the onset of HTN. UA also directly acts on endothelial cells, resulting in reduced nitric oxide levels. This affects vascular smooth muscle cell proliferation, extracellular matrix deposition, and macrophage adhesion and migration, which leads to arterial resistance and remodeling, followed by renal dysfunction and renal fibrosis (Fig. 2).

Hypothetical mechanisms of renal damage caused by hyperuricemia. NOS, nitric oxide synthase; RAS, renin-angiotensin system; COX-2, cyclooxygenase 2; VSMC, vascular smooth muscle cell; EMT, epithelial-to-mesenchymal transition.
Multiple recent studies have suggested that treatment of hyperuricemia may delay or prevent the onset of CKD. Feig et al. [60] conducted a randomized double-blinded study, which showed that treatment of hyperuricemia in adolescents with newly diagnosed HTN was effective in controlling BP. However, there is no evidence to support CKD treatment for asymptomatic hyperuricemia in patients with HTN. Although animal experiments suggest that UA causes non-crystalline and crystal deposition damage to the kidney, there is insufficient evidence for human patients. The results have differed among observational studies, and there is a lack of large-scale randomized controlled trials to confirm the benefits of UA reduction. The Febuxostat versus Placebo Randomized Controlled Trial Regarding Reduced Renal Function in Patients with Hyperuricemia Complicated by Chronic Kidney Disease Stage 3 (FEATHER) trial showed no positive results for febuxostat compared with a placebo, but this finding was only for a small number of subgroups without proteinuria and low serum creatinine [61]. Nonetheless, most studies suggest that UA-lowering therapy can delay CKD progression. A single-center double-blind, randomized, parallel placebo-controlled study indicated that, compared with a placebo, UA reduction was able to slow the decline of glomerular filtration rate in patients with stage 3 and 4 CKD [62]. Siu randomly divided 54 patients with both CKD and hyperuricemia into two groups. The experimental group was treated with allopurinol to reduce serum UA levels. At the 1-year follow-up, four of the 26 patients in the treatment group exhibited renal functional deterioration, whereas 46.1% of patients in the control group exhibited renal functional deterioration [63]. Although the results did not differ significantly between the two groups, the reduction of serum creatinine levels tended to be greater in the treatment group than in the control group. In another small-scale study, the investigators concluded that UA-lowering therapy reduced inflammation and slowed the progression of renal disease in patients with both CKD and hyperuricemia [64]. These studies’ findings suggest that UA reduction may improve renal function. The clinical trials of UA-lowering therapy are listed in Table 1 [61–71]. The question raised by the findings of these studies is whether the benefit of UA-lowering therapy is related to the reduction in UA itself or to the inhibition of XO. The results of two additional trials indicate that both benzbromarone and febuxostat could reduce the risk of CKD progression and lower serum UA levels in patients with CKD [65,66]. We suspect that the benefit of UA-lowering therapy mainly arises from the reduction in UA itself, although the inhibition of XO may amplify this effect and should be evaluated by credible experimental studies.

Randomized controlled trials of uric acid-lowering therapy for the recovery of renal function.
Although there has been increasing evidence to support hyperuricemia as a true risk factor for CKD, there remains no agreement regarding whether treatment of asymptomatic hyperuricemia with UA-lowering therapy will offer greater renal protection. Large trials are needed to clearly evaluate this issue.
HYPERURICEMIA AND CVD
CVD is a complex disease with multiple underlying mechanisms, for which hyperuricemia could be considered a contributing factor. Many studies have investigated the relationship between hyperuricemia and CVD. This relationship was originally established in the early 1900s. Elevated serum UA has been linked to an increased risk of HTN and CVD [72]. In those studies, serum UA levels were frequently elevated in patients with high BP, obesity, and IR. Higher levels of hyperuricemia were strongly associated with various macrovascular and microvascular diseases in a recent study of 15,773 participants, which revealed that the rates of mortality and cardiac mortality gradually increased with increasing levels of serum UA [73].
There remains debate regarding whether UA is an independent predictor of CVD. Hyperuricemia is presumed to be an independent risk factor for the incidences of both HTN and CVD. Many epidemiologic studies have confirmed an association between hyperuricemia and CVD, which has been further addressed in numerous prospective studies [3]. Notably, it appears more accurate to regard hyperuricemia as a consequence of the existence of previously related cardiovascular risk factors [74].
UA increases BP through endothelial cells and vascular smooth muscle cells [11]. Hyperuricemia can contribute to renal vasoconstriction and elevated BP levels by stimulating the renin-angiotensin system and inhibiting the release of endothelial nitric oxide [56]. A previous study of patients with HTN showed that when the serum UA levels exceeded 7.5 mg/dL in men and 6.2 mg/dL in women, the CVD risk increased significantly [75]. A subanalysis by Alderman [76] showed that a 1 mg/dL increase in serum UA level could potentially increase cholesterol levels by 20 mg/dL and elevate systolic BP by 10 mmHg. More common findings were reported in the Relation Between Serum Uric Acid and Risk of Cardiovascular Disease in Essential Hypertension (PIUMA) study: when serum UA levels exceeded 6.2 mg/dL in men and 4.6 mg/dL in women, the CVD risk increased significantly [77]. The National Health and Nutrition Examination Survey (NHANES) III study findings were similar, in that CVD risk increased significantly when the serum UA level exceeded 6.0 mg/dL in both sexes [78].
A large number of observational studies revealed that an increasing UA level was an independent risk factor for the risk of HTN. Among studies of CVDs other than HTN, a 3-year cohort study (J-CAD) was conducted in Japan, involving patients with coronary artery disease. The findings were similar to the NHANES III findings, in that an increasing risk of CVD events (including death) was observed in patients with serum UA > 6.8 mg/dL [79]. Serum UA has also been linked with the incidences of atrial fibrillation [80,81], coronary artery disease [82], and heart failure mortality [83]. Notably, in patients with both acute and chronic heart failure, morbidity, and mortality rates were significantly elevated concomitantly with serum UA levels [84].
The current evidence and studies demonstrated that approximately 40% of patients with heart failure exhibited elevated levels of serum UA. This was previously linked to pro-inflammatory heart failure and oxidative stress, which might have led to worsened clinical symptoms. Furthermore, a substantial proportion of patients with both heart failure and hyperuricemia reported renal functional decline. Most of these trials suggested that the cardiovascular effects associated with UA levels are related to its intracellular effects [85]. Thus, the use of XO inhibitors for treatment was presumed to be most potent in the reduction of intracellular UA because these agents block intracellular production and diminish extracellular levels. Furthermore, febuxostat (a nonpurine XO inhibitor) was confirmed to substantially reduce the levels of serum UA in patients with hyperuricemia [86]. However, this treatment involves a risk of CVD.
Sex differences regarding the associations of serum UA have been reported in multiple previous studies [87,88]. In particular, women of premenopausal age tend to have lower serum UA levels than men, presumably due to the uricosuric effects of estrogens [89]. Serum UA levels tend to increase in women of postmenopausal age, due to the influence of sex hormones [90]. These sex differences contribute to differences involving metabolic syndrome, CVD, and cancer [89].
Previous studies have analyzed the associations between serum UA and cardiovascular events in the context of sex differences involving serum UA. Some studies showed that the serum UA level is an independent risk factor for cardiovascular mortality, regardless of sex [91,92]. Other studies revealed a clear difference in cardiovascular mortality between men and women [93,94]. Finally, Niskanen et al. [95] reported that the level of serum UA is a strong predictor for cardiovascular mortality in middle-aged men. Thus, there remains debate regarding the effects of sex on the association between serum UA and cardiovascular mortality.
In a recent retrospective observational study in Gunma, Japan [96], 12,029 participants were enrolled to evaluate the association between UA level and HTN risk according to age and sex. The results showed that older men had lower UA levels, whereas older women had higher UA levels, compared with younger participants. Moreover, in men ≤ 50 years of age, there was a significant association between UA level and HTN incidence, but not in men ≥ 50 years of age. By contrast, in women ≥ 40 years of age, there was a significant association between UA level and HTN incidence, but not in women ≤ 40 years of age. These data suggest that UA level is an independent indicator of HTN among men ≤ 50 years of age and women ≥ 40 years of age.
In another recent observational study that assessed the sex-specific association between serum UA and all-cause mortality in 40-year-old patients [97], 27,490 participants were enrolled. The results showed that a lower serum UA level was an independent risk factor for all-cause mortality in men but not in women. Furthermore, serum UA levels were lower in women than in men. With increasing age, serum UA significantly increased in women and declined in men, consistent with the findings of other studies [88,98].
CONCLUSIONS
Hyperuricemia is a clinically important factor for the onset of CKD. Many studies have suggested that UA itself may harm patients with CKD by enhancing inflammation and promoting the progression of CKD. Additionally, a high serum UA level may cause kidney damage through a crystal-dependent pathway and non-crystal-dependent mechanisms such as inflammation, oxidative stress, and hemodynamic alterations. Most clinical studies have suggested that early treatment of hyperuricemia is beneficial for the control of CKD, HTN, and other chronic diseases. Because hyperuricemia represents a nontraditional risk factor for CKD, the close association between hyperuricemia and CKD requires more intensive inquiry. Hyperuricemia and the prevention of both CKD progression and cardiovascular events is an important research field, with large studies expected in the near future. Pre-existing CVD in patients with both hyperuricemia and CKD demands urgent attention. In particular, levels of serum UA should be carefully assessed in patients at high risk of CVD. Diet, polypharmacy, and lifestyle issues are very important aspects to discuss with patients, especially in countries where medications can be bought without a prescription, as UA can be altered by many medications. Special attention is needed for specific contraindications to certain drugs. Febuxostat is the drug of choice for the treatment of patients with hyperuricemia and is effective at preventing CKD progression, but its adverse effects on the cardiovascular system require careful attention. Although several studies focused on UA and CVD have been recently reported, future large-scale studies are expected.
Notes
No potential conflict of interest relevant to this article was reported.
Acknowledgements
We are grateful to Prof. Sun Dong our director of the postgraduate course, at the Xuzhou medical university, Xuzhou Medical Affiliated Hospital, Department of Nephrology for sharing his knowledge and guiding and caring, and our second commendations go to our dear collogues and the staff of our department with their great support and assistance.
This study was supported by funding from a project of Jiangsu Provincial Post Graduate Innovation Plan (SJCX17_0560).