Daclatasvir and asunaprevir combination therapy for patients with chronic hepatitis C virus genotype 1b infection in real world
Article information
Abstract
Background/Aims
Previous studies have reported a high rate of sustained virologic response (SVR) and a low rate of serious adverse events with the use of daclatasvir (DCV) and asunaprevir (ASV) combination therapy. We evaluated the efficacy and safety of DCV and ASV combination therapy for patients with chronic hepatitis C virus (HCV) genotype 1b infection in real world.
Methods
We enrolled 278 patients (184 treatment-naïve patients) from five hospitals in Daegu and Gyeongsangbuk-do. We evaluated the rates of rapid virologic response (RVR), end-of-treatment response (ETR), and SVR at 12 weeks after completion of treatment (SVR12). Furthermore, we investigated the rate of adverse events and predictive factors of SVR12 failure.
Results
The mean age of patients was 59.5 ± 10.6 years, and 140 patients (50.2%) were men. Seventy-seven patients had cirrhosis. Baseline information regarding nonstructural protein 5A (NS5A) sequences was available in 268 patients. Six patients presented with pretreatment NS5A resistance-associated variants. The RVR and the ETR rates were 96.6% (258/267) and 95.2% (223/232), respectively. The overall SVR12 rate was 91.6% (197/215). Adverse events occurred in 17 patients (7.9%). Six patients discontinued treatment because of liver enzyme elevation (n = 4) and severe nausea (n = 2). Among these, four achieved SVR12. Other adverse events observed were fatigue, headache, diarrhea, dizziness, loss of appetite, skin rash, and dyspnea. Univariate analysis did not show significant predictive factors of SVR12 failure.
Conclusions
DCV and ASV combination therapy showed high rates of RVR, ETR, and SVR12 in chronic HCV genotype 1b-infected patients in real world and was well tolerated without serious adverse events.
INTRODUCTION
Until 2011, the standard antiviral treatment used to treat patients with chronic hepatitis C virus (HCV) genotype 1 infection was a combination of pegylated interferon (PEG-IFN) plus ribavirin (RBV) [1,2]. Following the introduction of direct-acting antiviral agents (DAAs) in 2011, the cure rate of chronic HCV infection has shown a marked improvement over the years. The first-generation DAAs combined standard treatment for patients infected with HCV genotype 1 was a combination of DAAs with PEG-IFN and RBV for 24 weeks, and it was observed that a sustained virologic response (SVR) rate of 68% to 89% was achieved in treatment-naïve patients [3-5]. However, interferon-based treatment was associated with several limitations such as adverse events, low tolerance, and limited indications for use particularly in patients with comorbidities.
In 2014, a newly developed nonstructural protein 5A (NS5A) inhibitor, daclatasvir (DCV) (Daklinza, Bristol-Myers Squibb, New York, NY, USA) and a nonstructural protein 3/4A (NS3/4A) protease inhibitor, asunaprevir (ASV) (Sunvepra, Bristol-Myers Squibb) were introduced as combination therapy without PEG-IFN and RBV in Japan, and showed a high rate of SVR and low rate of serious adverse events [6-9]. This combination therapy has been used in Korea since August 2015 following insurance approval.
We report the efficacy and safety of DCV and ASV combination therapy for patients with chronic HCV genotype 1b infection in real world.
METHODS
Patients
We retrospectively evaluated patients diagnosed with HCV genotype 1b infection who received DCV and ASV combination therapy between August 2015 and March 2017 at five university hospitals in Daegu and Gyeongsangbuk-do. Exclusion criteria for this study were: patients diagnosed with decompensated cirrhosis, those with hepatocellular carcinoma (HCC) requiring treatment, those with a history of liver transplantation, and coinfection with other HCV genotypes, hepatitis B virus, or the human immunodeficiency virus. Decompensated cirrhosis was defined as cirrhosis categorized as Child-Turcotte-Pugh class B or C based on the results of blood tests and imaging studies such as abdominal ultrasonography and/or computed tomography. The enrolled patients were divided into three groups: the treatment-naïve, treatment failure (including null response, partial response, virologic breakthrough, relapse after previous treatment), and treatment-intolerant group (those who discontinued treatment owing to adverse events). The study protocol was approved by the Institutional Review Board (CR-17-021) of each participating hospital. Patients were not required to provide informed consent because this study was based on a review of medical records, and patients had consented to treatment prior to initiation of treatment.
Treatment regimen
The treatment regimen used in this study was based on the Korean Association for the Study of the Liver (KASL) guidelines [10]. DCV and ASV combination therapy was initiated at standard oral doses: a single DCV tablet of 60 mg administered once daily and an ASV soft gel capsule of 200 mg administered twice daily for 24 weeks. Based on KASL guidelines, the ASV dose was reduced to 200 mg once daily over the same period in patients with a glomerular filtration rate (GFR) < 30 mL/min.
Assessment of resistance-associated variants
Patients were examined to identify pre-existing resistance-associated variants (RAVs) to NS5A using commercially available direct sequencing assays—the Veriti 96 well thermal cycler and genetic analyzer (Applied Biosystems, Carlsbad, CA, USA) with the upper limit of quantitation set at 5,000 IU/mL. The result was recorded as positive or negative based on the presence of RAVs at positions L31 or Y93.
Clinical and laboratory assessment
Blood tests including the baseline complete blood cell count, serum aspartate aminotransferase (AST), alanine aminotransferase (ALT), total bilirubin, serum albumin, γ-glutamyl transferase, prothrombin time, blood urea nitrogen, creatinine, GFR, and HCV RNA titer were obtained every 4 weeks during administration of the DCV and ASV combination therapy, and 12 weeks after completion of treatment. The HCV RNA titer was measured at each hospital using a real-time polymerase chain reaction assay. In patients who consented to undergo the procedure, transient elastography (FibroScan, Echosens SA, Paris, France) was performed to measure liver stiffness at baseline and after completion of treatment. Additionally, the fibrosis-4 (FIB-4) index was calculated to compare between the baseline liver stiffness and that observed 12 weeks after completion of treatment in patients who achieved SVR12.
Assessment of efficacy and safety
Efficacy was assessed by measuring the virologic response. Virologic response was defined based on the KASL guidelines [10]. Rapid virologic response (RVR) was defined as undetectable HCV RNA levels after 4 weeks of treatment. End-of-treatment response (ETR) was defined as undetectable HCV RNA levels after 24 weeks of treatment. SVR12 was defined as undetectable HCV RNA levels (< 15 IU/mL) at the 12-week time point after completion of treatment. Safety was assessed by measuring the rate of occurrence of adverse events. The attending physicians at each hospital reviewed patients’ medical records to identify adverse events.
Statistical analyses
Data were expressed as mean ± standard deviation or number (percentage). Therapeutic efficacy was evaluated using a modified intention-to-treat analysis. The chi-square test and the two-sample t test were used to perform univariate analyses. A p value less than 0.05 was considered statistically significant. The SPSS statistical software version 21.0 for Windows (IBM Co., Armonk, NY, USA) was used for data analyses.
RESULTS
Baseline characteristics of patients
A flow chart outlining the study has been presented in Fig. 1. Our study included 278 patients with chronic HCV genotype 1b infection, who received DCV and ASV combination therapy. Patients’ baseline characteristics have been summarized in Table 1. The mean age was 59.5 ± 10.6 years, and 140 patients (50.2%) were men. Among 268 patients in whom information regarding baseline NS5A sequences was available, six (2.1%) showed positive NS5A RAVs. Among these patients, three were positive for Y93 and three for L31. Seventy-seven patients (27.6%) were diagnosed with compensated liver cirrhosis. Mean body mass index (BMI) was 23.6 ± 4.5 kg/m2. Forty-three patients (15.5%) reported history of heavy alcohol consumption.
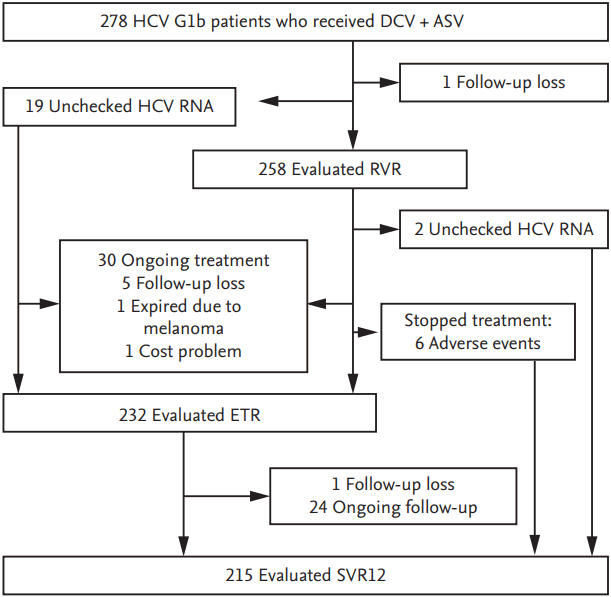
Flow diagram of the study. HCV, hepatitis C virus; DCV, daclatasvir; ASV, asunaprevir; RVR, rapid virologic response; ETR, end-of-treatment response; SVR12, sustained virologic response at 12 weeks after completion of treatment.

Baseline characteristics of enrolled patients
Among patients in whom treatment was initiated with DCV and ASV, 215 underwent final statistical analysis to evaluate the effectiveness and safety of this combination therapy. The remaining patients were excluded due to causes as follows: (1) ongoing treatment (n = 54), (2) lost to follow-up (n = 7), (3) expired secondary to malignant melanoma (n = 1), and (4) discontinued treatment secondary to the cost of treatment (n = 1). Among the 215 patients who were included in the analysis, 140 belonged to the treatment-naïve, 59 to the previous treatment failure, and 16 to the treatment-intolerant group.
Virologic response
The RVR and ETR rates were 98.0% (253/258) and 95.2% (221/232), respectively. We analyzed 215 patients for SVR12. The overall rate of SVR12 was 91.6% (196/215). Subgroup analysis revealed SVR12 rates of 90.7% (127/140) in the treatment-naïve, 91.5% (54/59) in the treatment failure, and 93.8% (15/16) in the treatment-intolerant group (Fig. 2). Among the 204 RAVs-negative patients, the overall rate of SVR12 was 91.7% (187/204). Subgroup analysis showed SVR12 rates of 91.0% (121/133) in the treatment-naïve, 92.8% (52/56) in the treatment failure, and 93.3% (14/15) in the treatment-intolerant group. We observed that three patients tested positive for RAVs (two for Y93, one for L31). Among these, one patient who was positive for L31 achieved RVR, but failed to achieve ETR and SVR12. The other two patients achieved RVR, ETR, and SVR12.
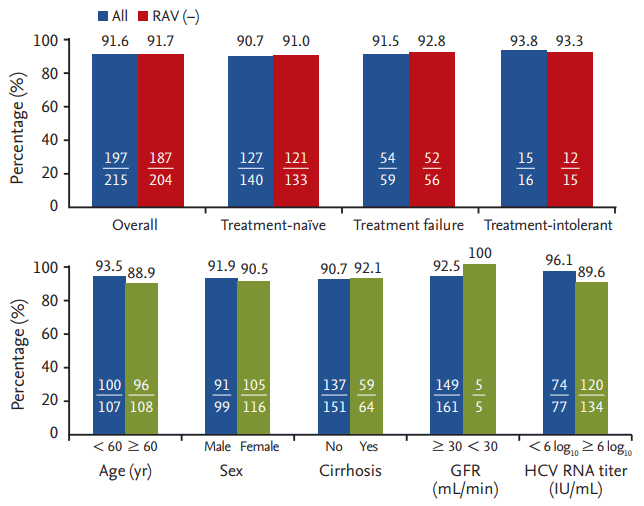
Rates of sustained virologic response at 12 weeks after completion of treatment (SVR12) according to baseline characteristics. RAV, resistance-associated variant; GFR, glomerular filtration rate; HCV, hepatitis C virus.
SVR12 was analyzed based on the patients’ baseline factors including age, gender, presence of liver cirrhosis, history of heavy alcohol consumption, presence of comorbidities such as diabetes mellitus and/or hypertension, BMI, positivity of RAVs, and HCV RNA titers. Univariate analysis did not show significant predictive factors of SVR12 failure (Table 2).

Univariate analysis of factors associated with failure of sustained virologic response at 12 weeks after completion of treatment
Adverse events
Adverse events noted in this study have been summarized in Table 3. Adverse events occurred in 17 patients (7.9%). Four patients complained of two symptoms. Six patients discontinued treatment because of adverse events. Four patients (1.9%) discontinued treatment because of liver enzyme elevation. The AST/ALT levels increased to > 5 times the upper limit of normal (ULN) at 8 weeks of treatment in one patient who belonged to the previous treatment failure group (null responder), and she failed to achieve SVR. Two patients belonging to the treatment-naïve group discontinued treatment because their AST/ALT levels increased to > 5 times the ULN at 8 and 12 weeks of treatment, respectively. However, they achieved SVR12 despite the short duration of treatment. One patient belonging to the treatment-naïve group who presented with underlying liver cirrhosis discontinued treatment because her AST/ALT levels increased to > 10 times the ULN, and she progressed to decompensated cirrhosis with jaundice and ascites at 11 weeks of treatment. Her liver function returned to the normal range following discontinuation of treatment. Regardless of this adverse event, she achieved SVR12. Of note, two patients (0.9%) discontinued treatment at 8 and 12 weeks of treatment, respectively because of severe nausea, which developed during the early period of treatment. These adverse events resolved after discontinuation of treatment. Eleven patients continued treatment despite the occurrence of adverse events. Five (2.3%) complained of fatigue, and four (1.9%) complained of headache. Other adverse events included diarrhea (n = 1), dizziness (n = 1), skin rash (n = 1), loss of appetite (n = 1), and mild dyspnea (n = 1).

Summary of adverse events (n = 216)
Improvement of liver function and liver stiffness after achievement of SVR12
We compared several biochemical markers in patients who achieved SVR12 (Table 4). Compared to baseline tests, we observed that AST, ALT, total bilirubin, and albumin were significantly improved at 12 weeks after completion of treatment (p < 0.05). Compared to baseline levels, liver stiffness (evaluated by transient elastography) showed significant improvement after completion of treatment (p = 0.026). Moreover, we compared the baseline FIB-4 index with that observed 12 weeks after completion of treatment. The mean baseline FIB-4 index was 4.38, which showed a marked decrease to 2.71 at 12 weeks after completion of treatment in those who achieved SVR12 (p < 0.001).

Changes in liver function and liver stiffness in patients who achieved a sustained virologic response at 12 weeks after completion of treatment
DISCUSSION
A previous study demonstrated that DCV and ASV combination therapy could achieve a SVR12 rate of 89.5% in Korean patients with HCV genotype 1b infection in real world [11]. In the present study, approximately 91.6% of patients with or without RAVs in NS5A achieved SVR12 after DCV and ASV combination therapy. The overall efficacy was observed to be marginally higher than in the previous study. Subgroup analysis in our study revealed SVR12 rates of 90.7% in the treatment-naïve, 91.5% in the treatment failure, and 93.8% in the treatment-intolerant group. Previous studies have reported that a history of previous treatment was a predictive factor for SVR12 [11,12]. However, in the present study, no significant difference was observed between subgroups in this regard. This observation could be attributed to the fact that only a few RAVs-positive patients were enrolled in our study.
Notably, previous studies showed that ALT and albumin levels, and the platelet count showed significant improvement after completion of treatment compared to baseline measurement in patients diagnosed with cirrhosis who achieved SVR12 [13]. Our study results are in agreement with previous reports. Additionally, we observed that the liver stiffness (measured using transient elastography and the FIB-4 index) showed significant improvement after completion of treatment. Eradication of HCV can ameliorate liver inflammation and liver fibrosis [14]. Mandorfer et al. [15] reported that DAA therapy decreases portal pressure in patients who achieved SVR who present with moderate hepatic fibrosis. This previous study supported the finding of improvement in liver function and liver stiffness in patients who achieved SVR in our study. The previous study reported that patients showing pretreatment viral loads of ≥ 6.0 log10 IU/mL demonstrated a significantly lower rate of SVR12 than that observed in patients showing pretreatment viral loads of < 6.0 log10 IU/mL [16]. The HALLMARK DUAL study reported the odds ratio of SVR12 was 3.38 for HCV RNA < 800,000 IU/mL. A comparison of the SVR12 rate between those with high versus low HCV RNA titers showed that the SVR12 rate was higher in those with a lower HCV RNA titer in treatment-naïve patients (96% vs. 87%, respectively), previous non-responders (93% vs. 80%, respectively), and in the ineligible/treatment-intolerant patients (88% vs. 80%, respectively) [17]. In our study, baseline higher viral loads of ≥ 6.0 log10 IU/mL were associated with a marginally lower rate of SVR12 than that observed in patients with pretreatment viral loads of < 6.0 log10 IU/mL (89.6% vs. 96.2%, respectively), although this result was not statistically significant (p > 0.05). This difference should be interpreted cautiously. Baseline characteristics of patients including age, gender, history of heavy alcohol consumption, presence of liver cirrhosis, comorbidities such as diabetes mellitus and/or hypertension, and BMI were not observed to influence SVR12 rates in our study.
DAA-based treatment is associated with relatively fewer adverse events than those noted with interferon-based treatment. Headache, fatigue, diarrhea, nausea, and nasopharyngitis are relatively common adverse events associated with DCV and ASV combination therapy [18]. Nasopharyngitis is observed to be the most common adverse event (29% to 36%), although we did not observe this adverse event in our study. Less common adverse events including bradycardia, pyelonephritis, atrial fibrillation, periarthritis, pneumonia, thrombocytopenia, hypochondriasis, and encephalopathy have been reported [9,12,19,20]. We did not observe any rare adverse events in this present study. A previous largescale study that discussed the use of DCV and ASV combination therapy reported that 2.9% of patients discontinued treatment because of liver-related events such as ALT elevation [21]. Another large-scale study revealed that discontinuation of combination therapy secondary to liver injury occurred in 28 of 924 patients (3.0%) [20]. In our study, the rate of discontinuation of therapy secondary to liver enzyme elevation was observed to be marginally lower than in previous studies. Reportedly, ASV can cause hyperbilirubinemia and elevated transaminase levels. Moreover, a few cases of immunoallergic hepatitis have been reported after the use of DCV and ASV combination therapy [22].
Limitations of our study are as follows. (1) This study was a retrospective study based on a review of medical records. Adverse events such as mild fatigue, general weakness or headache might not have been clearly described or might have been ignored depending upon the personalities of physicians or patients. (2) Only a few patients presenting with RAVs were enrolled in our study because DCV and ASV combination therapy is not approved by insurance for such patients in Korea. Therefore, even if treatment had been initiated in a patient prior to analysis and detection of RAVs, if the patient tested positive for RAVs, treatment was discontinued at that point. Thus, we could not obtain adequate data on a sufficient number of RAVs-positive patients to accurately evaluate the influence of RAVs on SVR failure. (3) We could not accurately assess the effect of DCV and ASV combination therapy on the prevention of progression to decompensated cirrhosis and the development of HCC in patients with cirrhosis because of the short study period.
In conclusion, DCV and ASV combination therapy showed high rates of RVR, ETR, and SVR12 when used to treat patients with chronic HCV genotype 1b infection in real world. DCV and ASV combination therapy showed good results regardless of experience of IFN-based treatment or baseline comorbidities. We observed that DCV and ASV combination therapy was well tolerated without serious adverse events. However, long-term follow-up of patients who achieved SVR12 after DCV and ASV combination therapy would be necessary to determine whether this treatment regimen can effectively prevent the development of HCC.
KEY MESSAGE
1. Daclatasvir and asunaprevir combination therapy showed high rates of rapid virologic response, end-of-treatment response, and sustained virologic response at 12 weeks after completion of treatment (SVR12) when used to treat patients with chronic hepatitis C virus genotype 1b infection in real world.
2. Daclatasvir and asunaprevir combination therapy was well tolerated without serious adverse events.
Notes
No potential conflict of interest relevant to this article was reported.