Diagnostic Usefulness of the Janus Kinase 2 Mutation in non BCR/ABL Myeloproliferative Disorders
Article information
Abstract
Background
We investigated the Janus kinase 2 (JAK2) mutation and its diagnostic value in patients suffering with non BCR/ABL myeloproliferative diseases (nMPD) or other reactive conditions.
Methods
We reviewed the clinical records of 83 patients who underwent bone marrow (BM) examinations with suspect of nMPD. The diagnoses of nMPD were made based on the WHO criteria since 2001 and the PVSG criteria before 2001. The JAK2 mutation was examined by PCR in 54 patients whose BM samples were available.
Results
The JAK2 mutation was detected in 25 patients (46%); 12 of 26 patients with essential thrombocythemia (ET), 9 of 12 patients with polycyhtemia vera (PV), one of 7 patients with chronic idiopathic myelofibrosis (CIM) and one patient with unclassifiable MPD. Additionally, JAK2 mutation was detected in each one patient with secondary polycythemia and reactive thrombocytosis. These two patients and two other patients among the JAK2 mutated ET did not meet the WHO PV criteria due to their initial low hemoglobin levels. These patients had liver cirrhosis and hypersplenism due to Budd-Chiari syndrome (1), gastrointestinal bleeding (1) or the initial hemoglobin level was slightly below the level as provided by the criteria, but the level showed a rising pattern despite cytoreductive therapy (2). With the results of the JAK2 mutation available, 4 patients' disease could be re-diagnosed as PV. Finally, the positive rate of the JAK2 mutation was 81% in PV, 48% in ET and 14% in CIM. The presence of JAK2 mutation closely correlated with PV (p=0.001), leukocytosis (p=0.001) and an increased cellularity of BM (p=0.024).
Conclusions
The JAK2 mutation may help differentiate nMPD from secondary cytosis. Therefore, it should be incorporated into the guidelines for the nMPD work-up for making a more accurate diagnosis and administering proper treatment.
INTRODUCTION
Myeloproliferative diseases (MPDs) are clonal hematopoietic stem cell disorders that are characterized by proliferation in the bone marrow (BM) of one or more of the myeloid (i.e. granulocytic, erythroid and megakaryocytic) lineages1). MPDs are traditionally classified into the "classic" and "atypical" subcategories2). The former category includes chronic myelogenous leukemia (CML), polycythemia vera (PV), essential thrombocythemia (ET) and chronic idiopathic myelofibrosis (CIM). The pathogenesis of CML is molecularly defined by a BCR/ABL rearrangement derived from t(9:22)(q34:q11)3). Another three entities are non-BCR/ABL-associated myeloproli-ferative diseases (nMPD) and clinicopathologically assigned disorders4-6). Both the formerly used diagnostic criteria of the Polycythemia Vera Study Group (PVSG)7) and the newly-developed World Health Organization (WHO) criteria are associated with difficulties in making the differential diagnosis or the individual categorization of nMPD and the differentiation of nMPD from reactive conditions8). In addition, the courses of PV, ET and CIM can vary, and these diseases require accurate diagnosis for appropriate management. The recent identification of the Janus kinase 2 (JAK2) point mutation (V617F) has provided the possibility of the molecularly defined diagnosis for PV, ET and CIM9-12). Therefore, we investigated the JAK2 mutation in patients who underwent BM examinations for the diagnosis of nMPD, and we evaluated correlation of the JAK2 mutation with the clinical factors associated with the diseases.
MATERIALS AND METHODS
We reviewed the clinical records of our patients who diagnosed as PV, ET and CIM according to the PVSG criteria before 2001 and the WHO criteria since 2001 as well as the secondary polycythemia (SP) or thrombocythemia (RT). Among these patients, we chose 54 patients whose cryopreserved BM samples were available for evaluation. Our study was approved by the institutional review board. The measurements of the red cell mass and the endogenous erythroid colonies (EEC) were unavailable for all the patients, and the serum erythropoietin results recently became available. Mononuclear cells from the BM were isolated using Histopaque-1077 (Sigma); DNA was extracted using the QIAmp DNA Blood Mini Kit (Qiagen). We used an 80 ng sample of each patient's DNA, which was amplified in a 36 cycles PCR reaction at an annealing temperature of 58℃. We used 10 µmol/L of the common reverse primer (5'- CTGAATAGTCCACAGTG TTTTCAGTTTCA-3') and 5 µmol/L of two forward primers. The first forward primer (5'- AGCATTTGGTTTTAAATTAGGAGTA TATT-3') was specific for the mutant allele, and it contained an intentional mismatch at the third nucleotide form the 3' end to improve the specificity (giving a 203 bp product): the second (5'- ATCTATAGTCATGCTGAAAGTAGGAGAAAG-3') primer amp lified a 364-bp product from both mutant and wild-type alleles, and it served as an internal PCR control, as was described previously9).
We compared the clinical characteristics at the time of the diagnosis and the vascular complications during the follow-up by using the chi-square test for gender, the diagnosis, splenomegaly, an increased M:E ratio, BM fibrosis or cellularity, low serum erythropoietin and vascular events both at the time of the diagnosis and during the follow-up, or we used the t-test for age and the levels of hemoglobin, the white blood cells (WBC) and platelets (PLT). The overall survival (OS) period was defined as the time from the diagnosis until death or to the last date the patient presented for follow up. The actuarial survival curves were plotted according to the Kaplan and Meier's method, and the survival differences, between the JAK2 mutation-positive and negative groups, were calculated using the log-rank test. All the calculations were performed with the SPSS system, version 13.0.
RESULTS
Patients' characteristics and clinical courses
Eighty-three patients underwent the BM aspiration procedure and biopsy due to suspected nMPD at the Gil Medical centers between November 1994 and February 2005. Cryopreserved BM mononuclear cells were available in 54 patients. These patients' characteristics, their treatment and their vascular events are summarized in Table 1. The underlying causes of SP were hepatoma (1) and Budd-Chiari syndrome (1). RT was associated with inflammation (3), bleeding or iron deficiency anemia (2) and herbal medication (1). After the average follow-up of 27 months (range 0~133 months), the median OS was not reached (Figure 1A). Six patients died at 1, 1, 4, 8, 9 and 12 months after the diagnosis, respectively. Their diagnoses were PV (1), ET (1), SP (2) and CIM (2). The main causes of the lethal outcome were infection in 4 patients with nMPD and hepatic failure in 2 patients with SP.
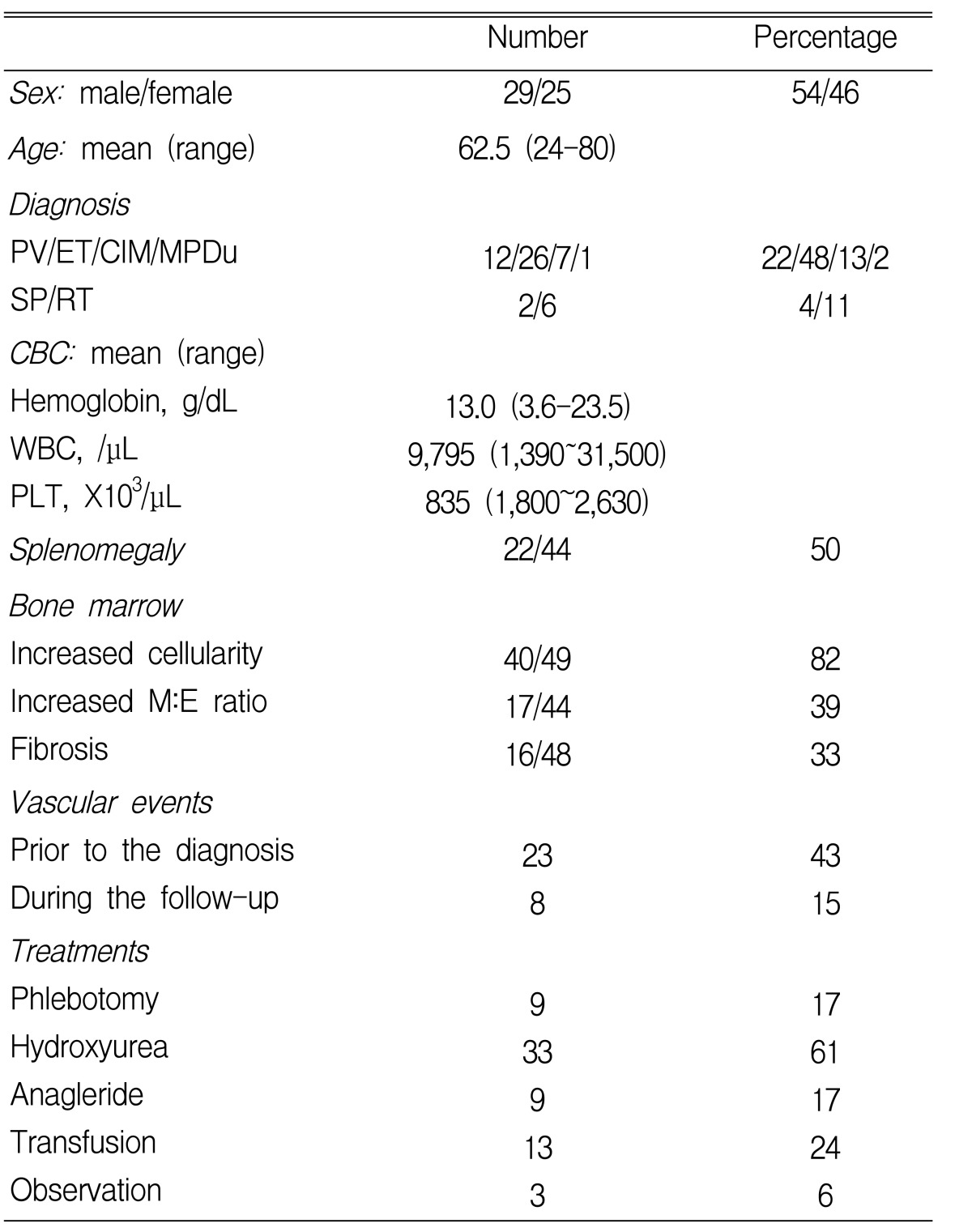
Patients' characteristics and clinical courses in 54 patients
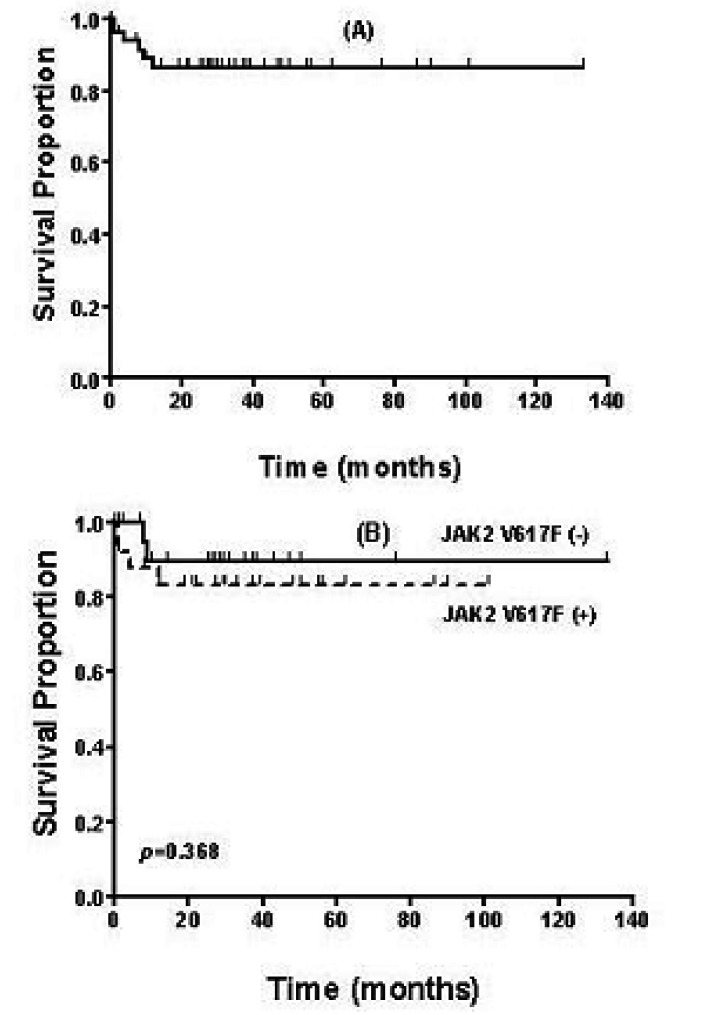
Overall survival in 54 patients (A) and according to the JAK2 mutation (B). No survival difference was noted between JAK2 V617F (+) and (-), p=0.368.
JAK2 mutation
Twenty-five patients (46%) showed a JAK2 mutation. These were 12 patients among the 26 patients with essential thrombocythemia (ET), 9 patients among the 12 patients with polycyhtemia vera (PV) and each of 7 patients with chronic idiopathic myelofibrosis (CIM), and 1 patient with unclassifiable MPD. Contrary to the initial expectations, the JAK2 mutation was established in each patient with secondary polycythemia and reactive thrombocytosis. These 2 patients with secondary cytosis and the other nMPD patients were closely re evaluated in respect of the accuracy of the initial diagnosis. When the clinical variables were compared according to JAK2 mutation, PV (p=0.001), leukocytosis (p=0.001) and the increased cellularity of BM (p=0.024) demonstrated a significant correlation with the presence of JAK2 mutation. However, hemoglobin, PLT counts, splenomegaly, BM fibrosis and vascular complications were not found to be related to the presence of the JAK2 mutation (Table 2). The OS periods were compared between the JAK2 mutation (+) and (-) patients, and no significant difference was noted (Figure 1B).

Clinical variables associated with the mutational status of JAK2.
Four cases with their diagnosis changed after the JAK2 mutation testing
Case 1 (SP → PV)
This 25 year-old woman visited our hospital in September 2002 with the complaints about a poor oral intake and abdominal discomfort. The abdominal examination revealed hepato-splenomegaly and ascites. Computer tomography of the abdomen showed obstruction of intrahepatic inferior vena cava. Insertion of two stents into hepatic vein was performed. The initial hemoglobin level was 14.2 g/dL, WBC 15,520/µL and PLT 375,000/µL. The BM aspiration and biopsy results showed an increased cellularity (80%) with a decreased M:E ratio (0.7). Iron staining revealed a depleted storage of iron. The initial diagnosis was Budd-Chiari syndrome with secondary polycythemia. During one year of the observation, her hematologic profile was within the normal range, except for leukocytosis (ranges: 12,33 0~28,550). The patient experienced recurrent episodes of spontaneous bacterial peritonitis, hepatic encephalopathy and hepatorenal syndrome. In September 2003 she was admitted to the emergency room and was found dead on arrival. We assume that the cause of the lethal outcome was hepatic failure aggravated by sepsis. After consideration of the JAK2 result, the patient's disease was re diagnosed as PV with a low hemoglobin level resulting from hypersplenism and irondeficiency anemia.
Case 2 (RT → PV)
This 37 year-old male patient came to our emergency room with a severe back pain in February 2002. The chest X-ray and cardiologic evaluation revealed no abnormality. Gastrofibroscopy showed an active duodenal ulcer. His hemoglobin level was decreased to 10.0 g/dL. The peripheral blood smear showed microcytic and hypochromic RBCs with a serum ferritin level of 3.63µg/L. The WBC was 14,690/µL and PLT 885,000/µL. The BM examination showed a slightly increased cellularity (60%) with a normal M:E ratio. His initial diagnosis was duodenal ulcer and iron deficiency anemia with reactive thrombocytosis. At his last visit on June 2003, his CBC profile showed: 15.6 g/dL of hemoglobin, 12,600/µL of WBC, 563,000/µL of PLT. After that, no further information on his hematologic change and clinical course has been available. After the JAK2 testing, we re-diagnose this patient's disease as PV associated with duodenal ulcer bleeding that caused transient low hemoglobin levels.
Case 3 & 4 (ET → PV)
A 72 years-old woman presented with gangrene of the second and third fingers of her right hand in May 1997. The CBC revealed 12.3 g/dL of hemoglobin, 26,500/µL of WBC and 1,254,000/µL of PLT. The RBCs were micorcytic and hypochromic. Gastrofibroscopy showed a scar of the gastric ulcer. The cellularity and megakaryocyte were increased. Her initial diagnosis was essential thrombocythemia. During the long period of the follow-up (101 months), her hemoglobin level increased to 15.1g/dL, and the PLT count dropped to 300,000/µL in February 2002, and the PLT maintained was around the lower limit of the normal. Only hydroxyurea was prescribed, and its dosage was reduced to 500 mg/day since February 2002. The PLT and WBC showed a decreasing tendency, while the hemoglobin level was increasing upto 16.5g/dL. With a JAK2 mutation, the diagnosis was changed to PV.
A 69 years-old woman was transferred to our hospital from a dental clinic in April 2001 due to an abnormal CBC. Her initial hemoglobin level was 16.4 g/dL, WBC 23,890/µL and PLT 1,116,000/µL. The patient had no symptoms or signs of bleeding. The BM aspiration and biopsy results showed an increased cellularity, megakaryocytes and an increased M:E ratio. The serum erythropoietin was below 1.7U/L. The CBC pattern was similar to that of the above mentioned 72 year old patient; the hemoglobin level was in excess of 16.5 g/dL twice over the period of 58 months. With a JAK2 result, her diagnosis was changed to PV.
DISCUSSION
Efforts to find the genetic changes that are responsible for developing MPD were recently brought to fruition with the discovery of the JAK2 mutation9-12). JAK2 is an upstream molecule that has been directly linked to erythropoietin receptor signaling. A short interfering RNA to prevent JAK2 expression could decrease the JAK2 protein levels, impair the spontaneous erythroid differentiation and markedly inhibit endogenous erythroid colony (EEC) formation13). On the basis of the above findings, all of the coding exons of the JAK2 gene were sequenced, and one G-to-T mutation at nucleotide 1849 in exon 12 was revealed. This leads to substitution of valine for phenylalanine at position 617(V617F)9, 10). The V617F mutation results in dysregulated JAK2 kinase activity: 1) this mutation is located in the JAK2 pseudokinase (JH2) domain, which negatively regulates activity of the kinase domain, 2) valine 617 and cysteine 618 are both important for maintaining JAK2 in an inactive conformation, 3) a mutation within the JH2 domain activates the kinase activity of the drosophila JAK homolog, and it results in a leukemia-like disorder14).
Until recently, the JAK2 mutation was detected at various frequencies in hematopoietic malignancies, and especially in MPD. The previously reported frequencies were 65~96% for PV, 23~57% for ET, 17~33% for CIM, 3~20% for chronic neutrophilic leukemia, 5% for the myelodysplastic syndrome and 0~2% for the hypereosinophilic syndrome9-12, 15-17). These findings imply that negativity of the JAK2 mutation does not mean seconday cytosis, and it shouldn't be used for excluding nMPD, and especially ET and CIM. Lymphoid malignancies did not show the JAK2 mutation18). In Korea, Lee et al reported a low incidence of 2.7% in acute myeloblastic leukemia19). There is no data on the JAK2 mutation in MPD for Oriental and Korean patients. When compared with the previously reported Western data, our data show a similar incidence of the JAK2 mutation for PV and ET, and its relatively low incidence in respect of CIM (Figure 2). We used the PVSG criteria of myelofibrosis from before 2001; thus, a portion of the prefibrotic or early fibrotic CIM in the WHO criteria might be classified as ET20). One person with the JAK2 mutation, among the 7 patients with CIM in our study, had a history of ET diagnosed 7 years earlier at another hospital. Although the initial data on this patient were not available, we could diagnose this patient's disease as CIM based on splenomegaly, bicytopenia and the dense fibrosis found on the BM study. The JAK2 mutation was established in one case of unclassified MPD. This 69 years old male had leukocytosis and hepato-splenomegaly. His BM examination revealed an increased cellularity without fibrosis. In another study, the JAK2 mutation was reported to be established in 54% (7/13) of the unclassified MPD patients16).

The rate of the positive JAK2 mutation in the disease categories after adjustment of the diagnosis in 4 patients.
The diagnosis of PV was associated with several practical difficulties before the introduction of the JAK2 testing. In Korea, measurements of the red cell mass and plasma volume are rarely available. If this study is available, then its diagnostic accuracy is indefinite despite the standardization of both the measurement technique and the interpretation criteria21). Therefore, the WHO criteria substitute the red cell mass for the absolute hemoglobin level: above 18.5 g/dL in men and 16.5 g/dL in women. In regard to the two PV patients with their hemoglobin levels lower than the above limit, Korean and Asian patients may have lower hemoglobin reference values than those noted in Western populations. With the data from performing regular screening of healthy persons, we must define the 99th percentile as our major criteria of PV. The EEC assay, a major criterion, and serum erythropoietin, a minor criterion, of the WHO standards were not performed in most patients in our study. The latter is now widely used in Korea, and it helps with making clinical decisions. With a value of < 3.3 U/L, a specificity of 97% with a positive predictive value of 97.8% were achieved for the diagnosis of PV22).
In our study, 4 cases of PV had been previously diagnosed as secondary polycythemia (1), reactive thrombocytosis (1) and ET (2). The case of secondary polycythemia strongly suggested the possibility of PV at presentation. This young woman presented with signs of Budd Chiari syndrome; therefore, the cause of her thrombophilic condition should be sought. Hypersplenism and combined IDA interfere with the exact diagnosis of PV. There was another report on three young female patients, who developed the Budd-Chiari syndrome as the first manifestation of PV, anf this strongly supports a routine screening for PV when thrombophilic conditions are found in young patients23). This approach can prove more useful with addition of the JAK2 mutation testing. Anemia with thrombocytosis could mask the presence of PV. These conditions are often confused with ET or reactive thrombocytosis. Among the 344 patients with erythrocytosis, 44 patients (13%) failed to initially conform to the diagnostic criteria of the WHO for PV because of their low hemoglobin levels24). One half of this group of patients (23/44) presented with thrombocytosis exceeding 600,000/µL; therefore, these findings suggested ET, but later these patients developed full-pattern PV as described in our two cases. Thus, screening for the JAK2 V617F mutation should be added as the next step after determining the serum erythropoietin in the diagnosis of PV, and especially when measurement of RCM is not available25).
In conclusion, the diagnosis of nMPD, including PV, can be more accurate or exclusive of reactive condition by testing for the JAK2 V617F mutation.