Alveolar Hemorrhage Associated with Crescentic Glomerulonephritis:
—A Case Report—
Article information
Abstract
Alveolar hemorrhage is a life threatening condition which requires an urgent diagnosis and treatment. We report a case of alveolar hemorrhage associated with crescentic glomerulonephritis without immune complex deposition with a review of the literature.
INTRODUCTION
Alveolar hemorrhage is a rare but life threatening condition, of which the fundamental process is diffuse bleeding into the acinar portion of the lung. The cardinal manifestations of alveolar hemorrhage include hemoptysis, alveola filling opacity on chest roentgenogram, anemia, dyspnea and hypoxemia. Alveolar hemorrhage is often misdiagnosed as pulmonary edema or pneumonia at its initial manifestation. Failure to diagnose and treat alveolar hemorrhage syndromes in their early stages can have two serious consequences, 1) alveolar hemorrhage may progress rapidly and lead to acute respiratory failure, or 2) alveolar hemorrhage is often an early manifestation of a systemic disease, which may subsequently damage extrapulmonary organs, particularly the kidney.
The major causes of alveolar hemorrhage are various, including anti-basement membrane antibody disease, i.e. Goodpasture’s syndrome; idiopathic pulmonary hemosiderosis; systemic lupus erythematosus and several systemic vasculitides. Since the pulmonary manifestations are similar to each other, etiologic diagnosis of alveolar hemorrhage syndrome is dependent largely on the extrapulmonary manifestation.
We have recently experienced a case of alveolar hemorrhage associated with cresentic glomerulonephritis without immune complex deposition on the immunofluorescence and electron microscopic examination of the kidney, lung and skin. We report the case with a review of the literature. There have been no reported cases of alveolar hemorrhage in Korean literature as far as the authors know.
CASE ROPORT
1. History and Physical Examination
A 68-year-old woman was admitted to Seoul National University Hospital because of hemoptysis and dyspnea, which developed 1 1/2 months and a week before admission, respectively. She had a history of blood-tinged sputum 30 years ago and it disappeared spontaneously without specific medical treatment. She was also exposed to mercury fumes at a plastic producing factory for two months 30 years ago, but there had been no specific problem. She was a non-smoker, non-alcoholic and there was no history of diabetes mellitus, hypertension, lung disease, renal disease, heart disease, frequent URI, or milk-allergy. One and a half months prior to admission, a blood clot was expectorated without dyspnea. She was admitted to another hospital and a chest roentgenogram at that time showed diffuse alveolar infiltration.
Diagnostic work-up, including transbronchial lung biopsy, revealed no specific diagnosis. She was referred to our hospital. During that period, hemoptysis persisted and four pints of packed cell were transfused. A week prior to admission, dyspnea developed and there were several episodes of paroxysmal nocturnal dyspnea.
On physical examination, she was dyspneic, but well oriented and alert. Vital signs were BP 100 mmHg/70 mmHg, pulse rate 83 times per minute, respiratory rate 30 times per minute and body temperature 36.3°C. The conjunctiva was slightly anemic and the sclera was not icteric. On chest auscultation, an inspiratory crackle was heard on the sides of both lower lung fields, there was no other abnormal finding.
2. Laboratory
At admission, a hemogram revealed: hemoglobin 8.0g/dl, hematocrit 237.7%, WBC 14500/mm3 and platelet count 497K. The erythrocyte sedimentation rate was 40 mm per hour. MCV, MCH and MCHC were within the normal range. Electrolytes were sodium 131 mEq/dl, potassium 5.37 mEq/dl and chloride 99mEq/dl. Arterial blood gas analysis revealed severe hypoxemia, pH 7.39, PaCO2 43 mmHg, PaO2 38 mmHg, and HCO3-26 mEq per deciliter at resting state. Blood urea nitrogen and creatinine were 21 mg/dl and 1.0 g mg/dl, respectively, and liver function tests were within the normal range. There was no bleeding tendency. Hepatitis B virus serology markers were all negative.
On urinary sediment examination, red blood cells were observed at a number of five to seven per high power field and the urine stick showed blood at the level of grade one. Ccr was 35 mg/dl/min. The Ham’s test, sucrose lysis test, Donath-Landsteiner test, and direct and indirect Coombs’ tests were all negative. The immunoglobulin level and complement level were checked twice during the hospital stay. The results on the seventh hospital day were IgG 1970 mg/dl (normal range 408–1788 mg/dl); IgA, 263 mg/dl (64–544 mg/dl); IgM 138 mg/dl (49–355 mg/dl); C3 95 mg/dl (62–212 mg/dl) C4 29 mg/dl (15–45mg/dl), and CH50 22.5 (150–250). The results of the second test on the 26th hospital day were IgG 2260 mg/dl; IgA 319 mg/dl; IgM 160 mg/dl; C3 51 mg/dl; C4 32 mg/dl and CH5050 11.1.
The chest roentgenogram showed diffuse alveolar opacity. (Figure 1) beginning from both the middle and upper lung fields and extending to the entire lung fields.
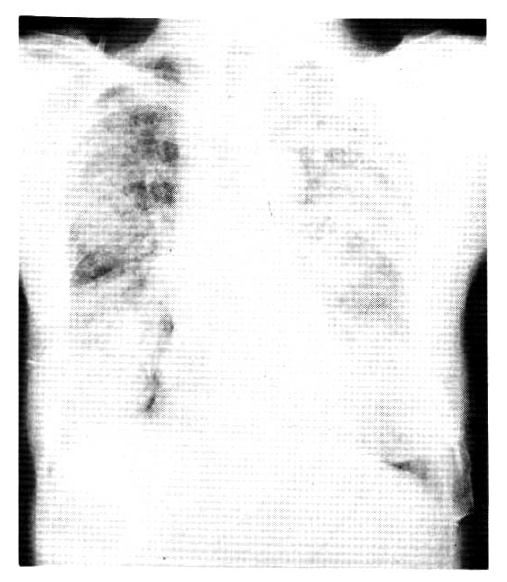
Chest roentgenogram before treatment.
The results of the resting pulmonary function test were forced vital capacity, 2.00 l/min and FEV1, 1.40 l/min; which suggests a mild restrictive and mild obstructive pattern. The exercise pulmonary function test suggested severe hypoxemia of pulmonary origin, arterial blood gas analysis at rest and exercise were pH 7.43; PaCO2 38 mmHg; PaO2 43 mmHg; bicarbonate 25 mEq/dl and pH 7.45; PaCO2 30 mmHg; PaO2 37 mmHg; bicarbonate 21 mEq/dl, respectively. DLCO was 97%. The value was falsely high, suggesting a recent pulmonary hemorrhage. Serum anti-GBM antibody by ELISA was negative. On bronchoscopy, bloody aspirate was drained from all bronchial openings. AFB staining was negative. Cytologic examination showed hemosiderin-laden macrophages. Bronchoscopic biopsy and transbronchial lung biopsy were not performed because of hemorrhage. Chest computed tomography and magnetic resonance imaging showed a bilateral diffuse air-space consolidation lesion without volume decrease.
The brain MRI showed small vessel ischemic changes. Under the impression of diffuse alveolar hemorrhage, open lung biopsy, percutaneous needle biopsy of the kidney and skin biopsy were performed to document the etiology.
3. Pathology Findings
Findings of the skin biopsy were normal and the lung specimen showed diffuse alveolar hemorrhage and hemosiderin-laden macrophages. There was also a moderate degree of interstial fibrosis. (Fig. 2 and 3). On immunofluorescence microscopic examination, there was no immune-complex deposition and basement membrane disruption was noted on electron-microscopic examination.
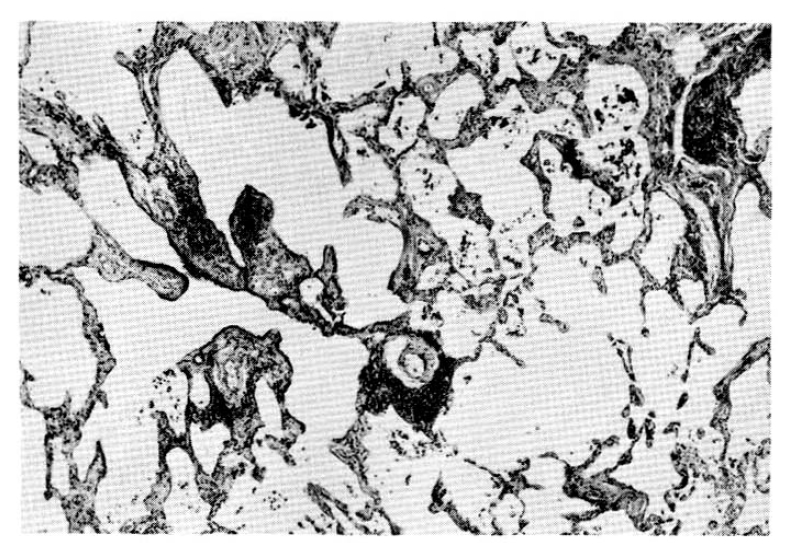
Photomicrograph of the lung biopsy, showing intersitial fibrosis and inflammation in the background of over-distended alveoli. Lymphoid cell collections are scattered together with patchy alveolar macrophage exudation. H & E × 40

Photomicrograph of the lung, showing collections of hemosiderin laden macrophages in the alveoli and also interstitial fibrosis. H & E × 100
The kidneys showed diffuse tubulointerstitial change and crescent formation on the glomerulus but no immune-complex deposition was noted on immunofluorescence microscopic examination and no electron dense deposit on electron microscope. (Fig. 4)

4. Therapy and Follow-up
Oral prednisolone 60 mg/day was given with INH prophylaxis from the 25th hospital day till the 41 st day. The chest X-ray showed an almost cleared lung parenchyme (Figure 5). There was no hemoptysis and the hemogram was Hb 9.8 g/dl, hematocrit 29.6%, WBC 6300/mm3 and platelet count 175K/mm3. Arterial blood gas analysis revealed pH 7.43, PaCO2 45 mrnHg, PaO2 59 mmHg and HCO3 30 mEq/dl.

Chest roentgenogram after oral prednisone therapy.
ON the 49th hospital day she was discharged and she appeared healthy on follow-up at the OPD. There was no further hemoptysis nor urinary abnormality.
DISCUSSION
Although there is no uniformly accepted scheme for classifying alveolar hemorrhage syndrome (AH), generally it includes five major categories; a) AH in anti-basement membrane antibody diseases b) AH in idiopathic pulmonary hemosidderosis c) AH in collagen vascular diseases and systemic vasculitides d) AH in idiopathic, rapidly progressive glomerulonephritis and e) AH secondary to exogenous agents (Table 1).

Alveolar Hemorrhage (AH) syndromes
The incidence of anti-basement membrane antibody disease in AH is 38% in one study, which comprises the most common form of alveolar hemorrhage syndrome.1)
The triad of hemoptysis, anemia and diffuse alveolar filling-opacities on chest roentgenogram suggests hemorrhage from the acinar portion, which accompanies hypoxemia and dyspnea. Careful diagnostic work-up should be performed, especially on the aspect of the kidney, skin, lung and the brain. Cutaneous manifestation suggestive of vasculitis may be of help. Synovitis, mononeuritis multiplex, abnormal urinary sediment and elevated creatinine level, allergy history, sinusitis and other extrapulmonary manifestations, all contribute to the diagnosis of the specific etiology of alveolar hemorrhage syndrome.
Bradley suggested a stepwise approach to the specific etiologic diagnosis of alveolar hemorrhage syndrome.4) The first step includes renal evaluation (urinalysis, BUN, creatinine), blood coagulation studies, study of sputum for microorganisms, serologic studies (anti-GBM antibodies, ANA, LE preparation, rheumatic factor, cryoglobulin, immune complexes), cardiac evaluation, visual inspection of the airway, and biopsy of lesions of the skin or airway. The second step, percutaneous needle biopsy of the kidney, should be performed if renal function is abnormal. Finally, if the specific etiology is unclear in spite of the previously mentioned studies, open lung biopsy might be performed. But considering the high morbidity and mortality of this procedure, therapy, often the pulse steroid therapy, might be instituted before open lung biopsy.
Leatherman summarized the differential diagnosis of alveolar hemorrhage syndromes.1) In the classification, AH associated idiopathic RPGN without immune complex deposition is described as a separate entity. Unlike Goodpasture’s syndrome, it has the following clinical features: Male to female ratio is 2:1, the mean age is 53 years showing side variation, and previous URI history is seen in 50% of the cases, renal biopsy shows glomerulonephritis features but the immunofluorescence finding is negative and extrapulmonary and extrarenal clinical manifestation is absent. The onset of alveolar hemorrhage is simultaneous with glomerulonephritis.
Leatherman determined the criteria of idiopathic RPGN without immune deposits as following2): (1) at least oen episode of pulmonary hemorrhage as manifested by hemoptysis and bilateral alveolar infiltrates on chest roentgenogram, (2) lack of evidence of more common causes of hemoptysis such as congestive heart failure, pneumonia, bronchitis, tuberculosis, pulmonary infarction, mitral stenosis or lung neoplasms, (3) normal results of coagulation studies and platelet counts, (4) presence of active glomerulonephritis by clinical and pathologic criteria, (5) absence of immunofluorescent features of anti-glomerular basement membrane antibody deposition in glomeruli and (6) absence of circulating anti-GBM antibody.
Stillmant also described the crescentic glomeruloneprhitis without immune deposits.8) He suggested the following features: (1) negative tests for circulating immune complexes and anti-GBM antibody, (2) evidence of acute and active disease documented by active urine sediments, increased protein excretion and normal kidney size as measured by i.v. pyelogram, renal arteriogram or examinatorial autopsy, (3) progressive loss of renal function as manifested by at least a twofold increase in serum creatinine concentration over baseline levels in a period of 1 to 3 months and (4) disease of less than 3 months duration as measured from the onset of symptoms or urinary abnormalities to the time of biopsy.
The present case is compatible with Leatherman’s criteria although the anti-GBM antibody test was performed by ELISA and the renal manifestation was not so dramatic as Stillmant described. When considering the immunoglobulin and complement activity of the case, it may be mediated by an immune mechanism which is different from Goodpasture’s syndrome. But small vessel ischemic changes of the brain cannot exclude vasculitis completely, which might manifest full-blown clinical features.
Therapy of alveolar hemorrhage syndromes is composed of oral steroids, cytotoxic agents including azathioprine or cyclophosphamide, antilymphocyte serum, anticoagulants, pulse steroids, plasma pheresis or combination.3) The most attractive form is the pulse steroid therapy which employs methylprednisolone in a dose of 30 mg/kg to a maximum single dose of 3g given intravenously over 20 minutes on alternate days for 3 days without diuretic therapy while under the careful monitoring of blood pressure, followed by alternate day oral prednisone 2 mg/kg in 48 hours. But the therapy is modified by specific entities; in Wegener’s granulomatosis, Cytoxan is preferred and in Goodpasture’s syndrome, plasma pheresis is suggested. We treated the patient with oral prednisone in a dose of 60 mg/day alone with marked improvement.
In summary, alveolar hemorrhage syndromes require urgent recognition, diagnosis of specific entities and application of therapeutic measures before the rapid and total course of the disease reaches the irreversible point.