Nasal Angiocentric Lymphoma With Hemophagocytic Syndrome
Article information
Abstract
Objectives
Hemophagocytic syndrome(HS) is a fatal complication of nasal angiocentric lymphoma (AL) and difficult to distinguish from malignant histiocyosis. Epstein-Barr virus(EBV)-associated HS is frequently observed in lymphoma of T-cell lineage and EBV is highly associated with nasal AL. Clinicopathologic features of 10 nasal ALs with HS were reviewed to determine the clinical significance and the pathogenetic association with EBV.
Methods
Ten patients of HS were identified from a retrospective analysis of 42 nasal ALs diagnosed from 1987 to 1996. Immunohistochemical study and in situ hybridization were performed on the paraffin-embedded tumor specimens obtained from 10 patients. Serologic study of EBV-Ab was performed in 3 available patients.
Results
Five patients had HS as initial manifestation, 3 at the time of relapse and 2 during the clinical remission of AL. Four patients were treated by combination chemotherapy (CHOP) and others had only supportive care. The median survival of all patients with HS was 4.1 months (range 2 days–36.5 months) and all had fatal outcome regardless of the treatment-modality. AH cases were positive for UCHL1 (CD45RO) and EBV by EBER in situ hybridization. The data of serologic tests indicated the active EBV infection.
Conclusions
HS is a fatal complication of nasal AL and has a high association with EBV. Reactivation of EBV may contribute to HS and further investigation of predictive factors and effective treatment of HS should be pursued in the future.
INTRODUCTION
Hemophagocytic syndrome (HS) is a systemic disease characterized by fever, pancytopenia, hepatosplenomegaly, lymphadenopathy, coagulopathy and histologically proliferation of histiocytes in the lymphoreticular system1, 2). It is usually associated with viral infection such as EBV, cytomegalovirus and adenovirus in the immunocompromised patients3, 4). Recently, it has been reported that EBV-associated HS is frequently observed in lymphoma of T-cell lineage and shows fatal outcome5, 6). It is difficult to distinguish HS from malignant histiocytosis (MH) and providing the suggestion of previously diagnosed MH may include HS in T-cell lymphoma. MH is a systemic malignancy derived from cells of the mononuclear phagocytic system and the essential diagnostic features include disseminated and progressive proliferation of morphologically atypical histiocytes and presence of phagocytosis by these neoplastic cells. HS has the similar clinical manifestations, but the cytologic atypia in hemophagocytic cells is not present7). Nasal angiocentric lymphoma(AL) has been classified as a peripheral T-cell lymphoma, but recently classified as nasal or nasal type T/NK cell lymphoma and highly infected with EBV8, 9). MH-like HS does not infrequently occur in nasal AL and usually has a rapidly fatal course8). We recently experienced a patient with nasal AL in whom, as a terminal event, a syndrome mimicking MH developed. In this study, we retrospectively analyzed AL with HS which had initially been suspected of having MH to assess the clinical significances and the pathogenetic association with EBV, and that would help to change our concept of MH and distinguish HS from MH.
MATERIALS AND METHODS
Patients
From 1987 to 1996, a total of 42 patients admitted to Catholic Cancer Center were established to have nasal AL. Ten of 42 patients showed HS during the clinical course of AL. The diagnosis of HS was based on a combination of the following clinicopathologic features: ① fever and splenomegaly, ② cytopenia of at least two hematopoietic series, and ③ over 2% of hemophagocytic histiocytes in the bone marrow1, 10). All patients underwent computed tomography of PNS and abdomen, plain chest X-ray and bone marrow examinations; they were staged according to the Ann-Arbor system11). The clinical and laboratory records were reviewed retrospectively.
Diagnosis of angiocentric lymphoma
The histopathologic diagnosis of AL was done by examination of paraffin-embedded tumor section with hemotoxylin-eosin stain. For immunophenotypical study, sections of the paraffin-embedded blocks were cut at 6 μm thickness and stained with specific monoclonal antibodies by the avidin-biotin complex (ABC) peroxidase methods described previously12). Mouse anti-human T-cell(CD45RO)(DAKO® Carpinteria, CA) and Mouse anti-human B-cell(IgG2a)(DAKO® Carpinteria, CA) were used for immunophenotyping of T and B lineage, respectively.
Association of angiocentric lymphoma with EBV
1) Serologic study of EBV-antibodies(Ab)
Serologic tests of IgG and IgM Abs against EBV-viral capsid antigen (VCA), early antigen (EA) and Abs against EBV nuclear antigens (EBNA) were performed in three available patients using the indirect immunofluorescence methods as previously described13).
2) In situ hybridization (ISH)
The EBV RNA in situ hybridization studies were performed in 10 patients using biotinylated 26bp oligonucleotides (Research Genetics) complementary to the most abundant early RNA sequence in EBV infections. 6 μm sections cut from paraffin block, placed on organosilane pretreated glass slides, were deparaffinized, dehydrated, predigested with pepsin solution and hybridized for 30 min at a concentration of 0.5 μg/ml of probe. After washing and blocking of endogenous peroxidase, detection was accomplished using streptavidin alkaline-phosphatase conjugate followed by development of signal with stable fast red TR/stable naphthol phosphate(Research Genetics) and counterstaining with hemotoxylin. A red color within the nucleus over background levels was considered as a positive reaction. A known EBV-positive neoplasm was a positive control and an EBV-negative lymphoid tissue was a negative control in each run.
Survival
The duration of overall survival was from the date of diagnosis of AL to the date of death or last follow-up. Survival duration from the diagnosis of HS was from the date of diagnosis to the date of death.
RESULTS
Patients’ clinical features
The basic clinical features and scheme of all patients were summarized in Table 1. There were 6 male and 4 female patients with median age of 43 years (range 23–63 years). The initial clinical stage of lymphoma were stage I (2 cases), II (3 cases), III (3 cases) and IV (2 cases). Five patients had HS as initial manifestation, three at the time of relapse of lymphoma, and two during the clinical remission of lymphoma. Four patients were treated with CHOP (cyclophosphamide, doxorubicin, vincristine and prednisone) chemotherapy and six had only supportive care. All cases showed rapidly fatal outcome and there was no significant difference depending on the methods of management. The median survival time from the diagnosis of HS of all patients, supportively cared patients and patients with CHOP, was 18 days (range 2–44 days), 18 days (range 2–27 days), and 19.5 days (range 14–44 days), respectively. The estimated median survival from the date of original diagnosis of AL was 4.1 months (range 2 days–36.5 months).
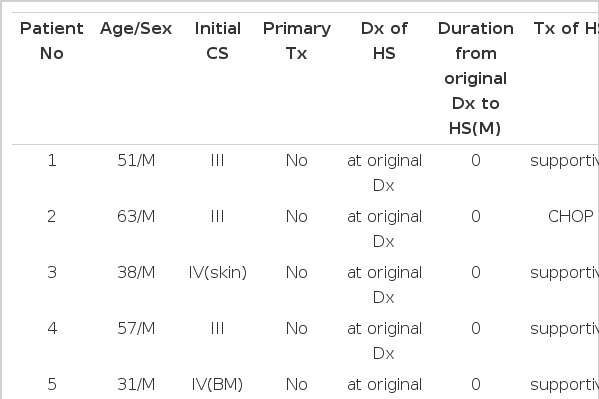
Patients’ clinical features
Histopathologic features
Polymorphic cellular composition was observed in six cases and the other 4 cases had relatively monomorphic composition. Angiocentricity was observed in 4 cases, and necrosis was in all cases. Immunophenotypical studies showed that all cases expressed T-cell phenotype. Representative histologic pictures of lymphoma and bone marrow of HS are shown in Fig. 1, 2, and 4, 5, respectively.
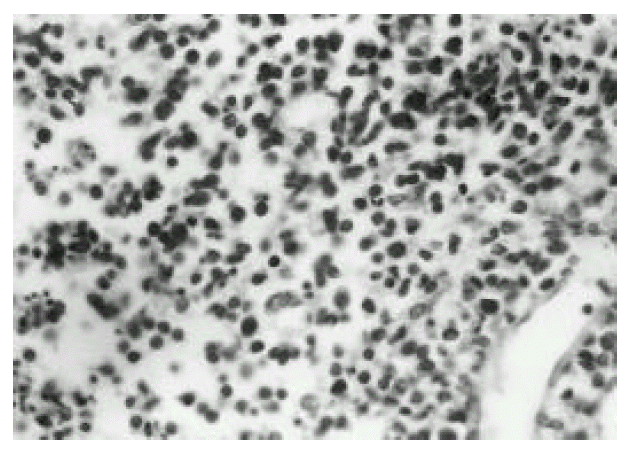
Medium-sized or large atypical cells with pleomorphic features and an occasional prominent nucleolus(H & E stain, × 200).

UCHL1 (CD45RO) expression on atypical lymphoid cells(Haematoxylin counterstaining, × 100).
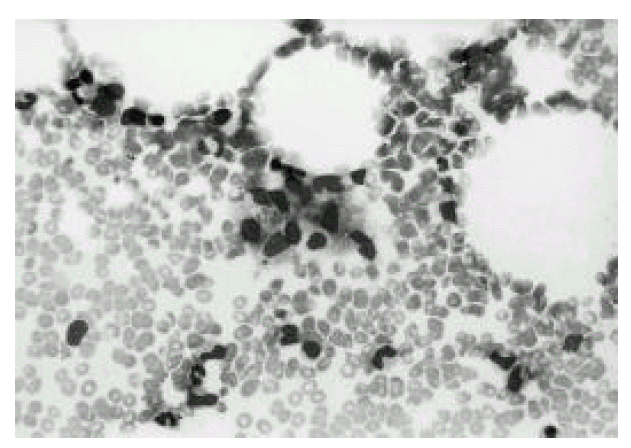
BM aspiration cytology shows increased histiocytes(arrow) (Wright stain, × 200).
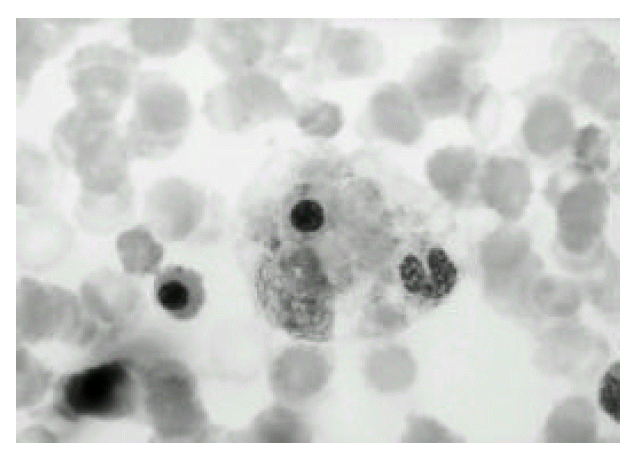
Histiocyte shows hemophagocytosis of normoblast & segmented neutrophil(wright stain, × 400).
Association of angiocentric lymphoma with EBV
The serological tests in three available patients were as follows; positive anti-EBNA Ab, positive anti-VCA IgG Ab(titer over 1:640), and positive anti-EA IgG Ab(1:320), which indicated the active infection of EBV.
In all cases, the EBV transcripts were seen in the nudei of the atypical cells by in situ EBER hybridization (Fig. 3).

Nuclear staining of the atypical lymphoid cells after EBER in situ hybridization(Haematoxylin counter-staining, × 200).
DISCUSSION
We recently experienced a patient with MH-like HS in nasal AL as a terminal event. HS has the similar clinical manifestations of MH and is difficult to distinguish from MH. Recently reassessment of patients previously diagnosed as having MH revealed that most cases were lymphoma of T-cell lineage or other, such as a virus-associated HS14). Retrospective analysis in our center revealed that 11 of a total 16 patients (from 1987 to 1996), with MH previously diagnosed, were MH-like HS. Nine of 11 patients, initially suspected as MH, were caused by nasal AL(data not shown).
Nasal AL which has the histologic feature of angiocentricity has been classified as a peripheral T-cell lymphoma but, recently, it is proving that it may include true natural killer (NK) cell lineage8, 9). Although angiocentricity is common to these tumors, it is not universally seen and the term AL has proved confusing. Therefore, it is proposed that the term ‘nasal or nasal type T/NK cell lymphoma’ should replace AL as a choice8). In this study, we only used pan T-cell marker(CD45RO) to distinguish from B-cell and all showed T-cell phenotype and it did not perform NK-cell phenotying due to lack of fresh specimens. More phenotypical studies including immunophenotyping and T-cell receptor gene rearrangement would be needed to distinguish the true T-cell lymphoma from NK-cell lymphoma. In this histologic study, necrosis and cellular polymorphism were a more common feature than angiocenricity and monomorphism, respectively.
HS was originally described in immunocompromised patients with viral infection3). Among various infectious agents, herpes virus, especially EBV, has been frequently implicated in the pathogenesis of VAHS3, 15). EBV is a ubiquitous Herpes virus with tropism for B-lymphocytes and oropharymgeal epithelium and has a strong association with endemic Burkitt’s lymphoma, B-cell lymphoproliferative lesions in immunocompromised patients and nasopharyngeal carcinoma13, 16). But recently, it appears that EBV has also been linked to about 40% of peripheral T-cell lymphoma and Hodgkin’s disease17–19). EBV is also more regularly detected in more than 80 % of nasal AL and, in these tumors, virtually all tumor cells harbour the virus. In this study, the presence of EBV was detected in all cases, particularly in most of the atypical lymphoid cells by the in situ hybridization, and the serologic test in three case indicated active infection. Although a causal relationship between EBV and AL is still undefined, the characteristic clinicopathologic features strongly suggest that EBV may contribute to the lymphomagenesis and the biologic features of AL and in situ hybridization and serologic study would be helpful for diagnosis and prediction of AL with HS8, 13) The mechanism of HS in AL is not fully understood yet, but it is thought to be caused by the cytokines, especially interferon-γ, tumor necrosis factor and interleukin-1 released from EBV infected lymphocytes20, 21). Further evaluation of the EBV-viral oncogenes and the microenvironment in HS would be needed to define the pathogenesis and the role of EBV in these tumors.
HS contributes to the high mortality of AL and usually has rapidly fatal outcome6, 22). It is difficult to predict HS in the course of nasal AL, but symptomatic recurrence and the histologic progression of the primary nasal lesion may be candidates. So, the repeated biopsy of the nasal lesion in symptomaticaly recurrent patients would be essential. In this study, most HS was developed in the active disease-status, but also even in the clinical remission of two cases. We could not confirm the postmortem pathologic staging of all cases due to lack of the autopsy-specimen. However, Jaffe et al.7) had reported that malignant lymphoma with erythrophagocytosis simulating MH had prevalence of lymphoma-involvements in lymph nodes, spleen, liver, lung, skin and kidney and hepatosplenomegaly was a common feature in all cases of HS, even in the patients with clinical remission. Therefore, pathologic staging, including laparoscopic biopsy, would be needed for the exact staging of lymphoma with MH-like HS.
There is no effective treatment for HS and, in literatural review, the combination of high-dose intravenous immunoglobulin and etoposide or high dose steroid were effective in some cases of HS2, 6, 23). Smith et al.24) described a young patient successfully treated with CHOP chemotherapy in the early phase of the clinical course. But, in this study, all patients showed as rapidly progressing into the fulminant course despite CHOP chemotherapy or palliative steroid pulse therapy. It seems that more aggressive treatment is needed in this condition and further investigation of the pathogenesis and biology of AL with HS should be pursued in order to improve the prognosis.