Protective Effect of Chlormethiazole, a Sedative, against Acetaminophen-Induced Liver Injury in Mice
Article information
Abstract
Objectives
The hepatotoxicity of acetaminophen is not a result of the parent compound but is mediated by its reactive metabolite N-acetyl-p-benzoquinone imine. Cytochrome P4502E1 (CYP2E1) is the principal enzyme of this biotransformation, which accounts for approximately 52% of the bioactivation in human microsomes. Recently, chlormethiazole, a sedative drug, is reported to be an efficient inhibitor of CYP2E1 activity in human beings. In this study we wished to evaluate whether chlormethiazole, an inhibitor of CYP2E1, could prevent acetaminophen-induced liver injury in mice.
Methods
Acetaminophen, at doses ranging from 200 to 600 mg/kg, was injected into the peritoneum of female C57BL/6 inbred mice fasted for four hours. Chlormethiazole (60 mg/kg) or 5% dextrose water was given 30 min before or 2 h after acetaminophen. Serum aminotransferase activities, histologic index score, survival rate and hepatic malondialdehyde levels were compared.
Results
Pretreatment with chlormethiazole 30 min before 400 mg/kg of acetaminophen completely inhibited acetaminophen-induced liver injury (median 118.5 U/L, range 75 to 142 vs. 14070 U/L, range 5980 to 27680 for AST; 49 U/L, range 41 to 64 vs. 15330 U/L, range 13920 to 15940 for ALT). In mice receiving chlormethiazole 2 h after acetaminophen, the mean AST and ALT levels were also less elevated, reaching only 20% of the value of acetaminophen-only group. These protective effects were confirmed histologically. Whereas more than 50% of mice died at 500 mg/kg of acetaminophen, all the mice pretreated with chlormethiazole survived at the same dose.
Conclusion
Chlormethiazole effectively reduces acetaminophen-induced liver injury in mice. Further studies are needed to assess its role in humans.
INTRODUCTION
Acetaminophen, a widely-used analgesic, is known to cause lethal hepatic necrosis at high doses. This acetaminophen-induced hepatotoxicity is not a result of the parent compound but is mediated by its reactive metabolite N-acetyl-p-benzoquinone imine (NAPQI)1). After low acetaminophen doses, NAPQI is inactivated by conjugation to glutathione2, 3). However, in acetaminophen overdose, or other circumstances which lead to depletion of glutathione2), the reactive metabolite binds covalently to hepatic proteins, which may initiate the development of hepatic necrosis4–7).
In humans, three cytochrome P450 enzymes, cytochrome P4502E1 (CYP2E1), cytochrome P4501A2 (CYP1A2) and cytochrome P4503A4 (CYP3A4), have been known to be involved in acetaminophen activation8–10), and most (30–78%) of this biotransformation is mediated by CYP2E18). Recently, it was reported that cyp2e1 knockout mice are resistant to acetaminophen-induced hepatic necrosis11). Also, many drugs or chemicals, which inhibit CYP2E1 activity, are known to prevent acetaminophen-induced liver injury in mice or rats12–15). However, most of these chemicals have limited data on safety in human beings.
Chlormethiazole is a sedative and anticonvulsive drug widely used in the treatment of alcohol withdrawal in Europe. Recently, it was reported that chlormethiazole inhibits not only the CYP2E1 transcription but also the catalytic activity of the enzyme in humans16).
Therefore, we investigated the effect of chlormethiazole on acetaminophen-induced liver injury in mice.
MATERIALS AND METHODS
1. Experimental Treatment of Animals
Female C57BL/6 mice (15–20 g body weight) were fed a standard diet up to 4 h before the experiment. Only drinking water was then allowed until 1 h after administration of acetaminophen, when food was reinstated. Acetaminophen (Sigma Chemical Co., St. Louis, Mo.) was dissolved in 40°C physiologic saline (25 mg/mL) and injected to the mice at doses ranging from 200 to 600 mg/kg intraperitoneally. Chlormethiazole was dissolved in 5% dextrose water (8 mg/mL) and administered at a dose of 60 mg/kg intraperitoneally. Each group in this study consisted of eight mice.
2. Serum Aminotransferase Determinations and Histologic Examination
Six groups of mice were studied: Group 1, mice were given equivalent volume of 5% dextrose water and normal saline to obtain control samples; Group 2, 5% dextrose water 30 min before 400 mg/kg of acetaminophen; Group 3, chlormethiazole 30 min before 400 mg/kg of acetaminophen; Group 4, 400 mg/kg of acetaminophen and then chlormethiazole 2 h after acetaminophen injection; Group 5, 5% dextrose water 30 min before 500 mg/kg of acetaminophen; Group 6, chlormethiazole 30 min before 500 mg/kg of acetaminophen.
24 h after acetaminophen injection, blood was collected for determination of serum aminotransferase activities and the mice were killed. The livers were immediately removed and sections of the livers were fixed in 10% formalin and embedded in paraffin for histologic examination.
Serum aspartate aminotransferase (AST) and alanine aminotransferase (ALT) were determined by the method of Reitman and Frankel with kits obtained from Sigma Chemical Company17).
Paraffin sections were stained with hematoxylin-eosin, according to standard procedures. Coded histologic sections were examined under light microscopy by a blinded observer. The extent of necrosis was graded from 0 to 4+ as follows: Histologically normal sections were graded 0. Minimal centrilobular necrosis was graded 1+; more extensive necrosis confined to centrilobular regions was graded 2+; necrosis extending from central zones to portal triads was graded 3+; and massive necrosis of most of the liver was graded 4+18).
3. Survival
In a separate experiment, 5% dextrose water and acetaminophen at doses ranging from 200 to 600 mg/kg were injected into mice of 5 groups. In another 4 groups of mice, chlormethiazole and acetaminophen at doses ranging from 200 to 500 mg/kg were injected. Animals were observed for 48 hours and the number of surviving animals at 48 h was counted. Two separate experiments with the same protocol were performed.
4. Hepatic Malondialdehyde Determinations
Three groups of mice were studied: Group 1, mice were given 5% dextrose water and normal saline; Group 2, 5% dextrose water 30 min before 400 mg/kg of acetaminophen; Group 3, chlormethiazole 30 min before 400 mg/kg of acetaminophen. Mice were sacrificed 6 h after acetaminophen administration. The livers were immediately removed and homogenized in ice-cold 1.15% (w/v) KCl solution. This homogenate was used to measure the extent of hepatic lipid peroxidation by the thiobarbituric acid method as described by Ohkawa et al19).
5. Statistical Analysis
Results are expressed as median with range. Nonparametric statistical procedures were used, and the significance of differences between groups was evaluated using the Mann-Whitney U test.
RESULTS
1. Serum Aminotransferase Activities
In mice receiving acetaminophen only, there was a striking elevation of serum transaminases (median 14070 U/L, range 5980 to 27680 for AST; median 15330 U/L, range 13920 to 15940 for ALT) at a dose of 400 mg/kg. Pretreatment with chlormethiazole 30 min before 400 mg/kg of acetaminophen completely inhibited acetaminophen-induced liver injury (median 118.5 U/L, range 75 to 142 for AST; median 49 U/L, range 41 to 64 for ALT). Furthermore, in animals receiving chlormethiazole 2 h after acetaminophen, the mean AST and ALT levels were less elevated (median 718 U/L, range 378 to 1191 for AST; median 3240 U/L, range 800 to 4260 for ALT), reaching only 20% of the value of the acetaminophen-only group (p < 0.001). Significant but less marked protection was observed when acetaminophen was injected at a dose of 500 mg/kg (median 857 U/L, range 380 to 3220 vs. 15560 U/L, range 3880 to 25540 for AST, p < 0.001; median 3462 U/L, range 1136 to 5132 vs. 14970 U/L, range 12110 to 20420 for ALT, p < 0.001, Fig. 1).

The effect of chlormethiazole on serum alanine aminotransferase (panel A) and aspartate aminotransferase (panel B) activity in mice treated with acetaminophen. Each group consisted of eight animals. Six groups of mice were studied: Controls, mice were given equivalent volume of 5% dextrose water and normal saline; AAP (400 mg/kg), 5% dextrose water 30 min before 400 mg/kg of acetaminophen; AAP (400 mg/kg) + CMZ 30 min pre, chlormethiazole 30 min before 400 mg/kg of acetaminophen; AAP (400 mg/kg) + CMZ 2 h post, 400 mg/kg of acetaminophen and then chlormethiazole 2 h after acetaminophen injection; AAP (500 mg/kg), 5% dextrose water 30 min before 500 mg/kg of acetaminophen; AAP (500 mg/kg) + CMZ 30 min pre, chlormethiazole 30 min before 500 mg/kg of acetaminophen. The horizontal bars correspond to the median value.
Abbreviations: AAP, acetaminophen; CMZ, chlormethiazole.
*P < 0.01 vs. AAP (400 mg/kg) group.
**P < 0.01 vs. AAP (500 mg/kg) group.
2. Histologic Changes
Typical extensive perivenular necrosis was observed in mice receiving acetaminophen only. Nearly all mice pretreated with chlormethiazole 30 min before 400 mg/kg of acetaminophen had histologically normal livers (p < 0.001). Mice treated with chlormethiazole 2 h after acetaminophen also had less severe necrosis than those receiving acetaminophen only (Fig. 2).

The effect of chlormethiazole on histologic index score in mice treated with acetaminophen. Grade 0: no damage; grade 1+: minimal centrilobular necrosis; grade 2+: more extensive necrosis confined to centrilobular regions; grade 3+: necrosis extending from central zones to portal triads; grade 4+: massive necrosis of most of the liver.
*P < 0.01 vs. AAP (400 mg/kg) group.
**P = 0.032 vs. AAP (500 mg/kg) group.
3. Survival
Fig. 3 shows the survival rate at doses ranging from 200 to 600 mg/kg of acetaminophen. Whereas more than 50% of mice died at 500 mg/kg of acetaminophen, all the mice pretreated with chlormethiazole survived at the same dose.

The effect of chlormethiazole pretreatment on the survival rate at various doses of acetaminophen. Each group consisted of eight mice. Two complete and independent experiments were performed. (▲) Acetaminophen group, and (•) chlormethiazole pretreatment group.
4. Hepatic Malondialdehyde Concentrations
There was no mouse with elevated hepatic malondialdehyde level in the group pretreated with chlormethiazole (median 39.7 pmole/mg protein, range 32.4 to 47.4), compared with those of controls (median 39.4 pmole/mg protein, range 35 to 51.9). Elevated levels were observed in about half of the mice treated with acetaminophen only (median 44.5 pmole/mg protein, range 35.1 to 109.6). However, the differences were not significant, statistically (0.25 < p < 0.5, Kruskal-Wallis one-way ANOVA by ranks, Fig. 4).
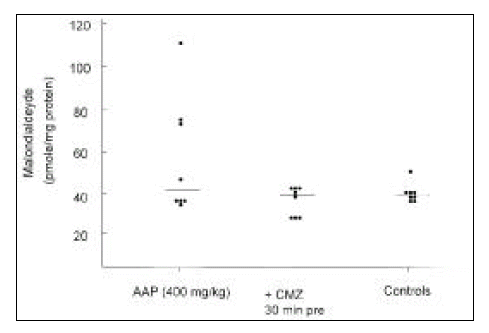
The effect of chlormethiazole on hepatic malondialdehyde level in mice treated with 400 mg/kg of acetaminophen. The malondialdehyde level was measured 6 h after acetaminophen administration.
DISCUSSION
Acetaminophen, a widely used analgesic, is known to cause potentially lethal hepatic necrosis. Hepatotoxic effect is expected in nonalcoholic patients with doses higher than 7.5 g. An overdose of 13 to 25 g is often lethal20). However, acetaminophen hepatotoxicity can occur in some individuals who ingest quantities within the therapeutic range. Alcoholics, with their induced cytochrome P-450 activity (especially, CYP2E1), attendant malnutrition and low glutathione levels, are predisposed to the toxic effects of acetaminophen. Prescribed doses as low as 4 g/d for 2 days have been associated with hepatotoxicity, and long-term use of as little as 2.6 to 3.9 g/d can result in significant hepatotoxic injury in alcoholics21). Concomitant use of isoniazid or barbiturate is also associated with increased hepatotoxic effect22).
Approximately 75% of an administered therapeutic dose of acetaminophen undergoes sulfation or glucuronidation; 5% to 10% is oxidized by the cytochrome P-450 enzymes to a toxic metabolite, N-acetyl-p-benzoquinone imine. Normally, N-acetyl-p-benzoquinone imine is conjugated with glutathione and safely excreted. With an overdose, increased oxidation occurs, glutathione becomes depleted and toxic N-acetyl-p-benzoquinone imine causes hepatocellular injury23). In humans, three cytochrome P450 enzymes, cytochrome P4502E1 (CYP2E1), cytochrome P4501A2 (CYP1A2) and cytochrome P4503A4 (CYP3A4), have been known to be principal catalysts of acetaminophen activation, and most (30–78%) of this biotransformation is mediated by CYP2E18–10).
As discussed above, cyp2e 1 knockout mice are known to be resistant to acetaminophen-induced hepatic necrosis11). Many drugs or chemicals that inhibit CYP2E1 activity are known to prevent acetaminophen-induced liver injury in mice or rats12–15). Cimetidine has been shown to prevent acetaminophen-induced hepatocellular injury in rats. However, the dose given in that experiment was 120 mg/kg, 18 times higher than the usual therapeutic dose in humans18). Additionally, studies performed with human microsome suggested a need for 5–10 times higher cimetidine concentrations than putative therapeutic concentrations24). Subsequent clinical trials have failed to prove the efficacy of cimetidine against acetaminophen-induced hepatic injury in humans20, 25). Other chemicals, such as cobalt chloride, methoxsalen or propylene glycol have been shown to prevent acetaminophen-induced hepatic injury in animal experiments, but most of these chemicals have limited data on safety in human beings12–15).
In the present study, chlormethiazole attenuated acetaminophen-induced hepatic injury in mice. However, it could not completely prevent hepatic injury at higher doses of acetaminophen. Other P-450s, such as CYP1A2 or CYP3A4 having a higher Km for acetaminophen, may be responsible for the toxicity at high doses of the drug.
Chlormethiazole was reported to inhibit CYP2E1 transcription as well as CYP2E1 catalytic activity in humans16). However, one study performed in vivo in rats showed no significant effect of chlormethiazole on CYP2E1 catalytic activity, but showed selective transcriptional suppression of CYP2E126). Therefore, the rat model seems to be not suitable to evaluate the therapeutic efficacy of chlormethiazole in acute acetaminophen-induced hepatic injury. It is not known whether chlormethiazole inhibits CYP2E1 catalytic activity at the constitutive level in mice. However, it can be speculated that chlormethiazole probably inhibits the CYP2E1 catalytic activity in mice because our data showed its protective effect against acute acetaminophen-induced hepatic injury. If chlormethiazole merely inhibits the CYP2E1 transcription, it is unlikely that chlormethiazole can nearly abolish hepatic injury in acute acetaminophen poisoning. Further studies are needed to confirm this assumption.
The dose of chlormethiazole given in this study produced mild sedation without respiratory depression in mice. At this dose, chlormethiazole nearly abolished acetaminophen-induced liver injury in mice. Since it was reported that a single administration of 192 mg of chlormethiazole in healthy human controls dramatically inhibits CYP2E1 catalytic activity16), the dose needed to prevent acetaminophen-induced liver injury may be within the usual therapeutic dose (1.5 g/d) in humans.
Lipid peroxidation has been observed after acetaminophen administration in vivo and in vitro in mice and rats27, 28). Although elevated levels were observed in about half of mice treated with acetaminophen, hepatic malondialdehyde levels among experimental groups in this study were not different statistically. The discrepancy between data from our study and previous studies may result from different doses of acetaminophen and different duration of starvation before acetaminophen administration. However, the possibility that lipid peroxidation may not be prerequisite for acetaminophen-induced hepatic injury cannot be excluded because mice with normal hepatic malondialdehyde level in the acetaminophen group also showed gross and histological evidence for hepatic damage. Other studies also have showed the late appearance of products of lipid peroxidation29).
Theoretically, chlormethiazole may be useful in preventing the CYP2E1-mediated generation of toxic intermediates. A very recent study has shown the protective role of chlormethiazole against ethanol mediated liver damage30), and our study also showed the protective role against acetaminophen-induced liver injury. Because chlormethiazole inhibits the generation of toxic metabolites, it is unlikely that chlormethiazole can substitute for N-acetylcysteine, which replaces the depleted glutathione stores necessary to prevent accumulation of toxic metabolite23), in acetaminophen overdose. In clinical practices, treatment with N-acetylcysteine within 10 hours of acetaminophen usually prevents severe liver damage. Severe hepatic necrosis usually develops when treatment is delayed31). So it is unlikely that patients who cannot benefit by N-acetylcysteine can benefit from chlormethiazole. Chlormethiazole may be useful in patients who cannot tolerate N-acetylcysteine side-effects such as vomiting or anaphylactoid reaction, or as a combination therapy with N-acetylcysteine. It may also be useful in situations when inhibition of CYP2E1 activity is beneficial. Administration of chlormethiazole before acetaminophen in alcoholic patients may be an example.
In conclusion, we have shown that chlormethiazole effectively reduces acetaminophen-induced liver injury in mice. Further studies are needed to assess its role in humans.
Acknowledgment
This study was supported by a grant from Ewha Womans University Mokdong Hospital.