Targeting immune dysregulation in chronic spontaneous urticaria: beyond antihistamines
Article information

Abstract
Chronic urticaria (CU) is characterized by recurrent wheals and angioedema lasting longer than six weeks. It affects approximately 0.5–1.4% of the population and profoundly impairs the quality of life. The disease exhibits a variable course, with many patients experiencing symptoms for several years. Higher disease activity correlates with increased burden and delayed remission. Therefore, early and effective intervention is critical to improving patient outcomes. Current first-line therapies target histamine, the principal effector mediator released from activated mast cells, using non-sedating H1-antihistamines. However, mast cells secrete a wide array of additional mediators, including platelet-activating factor, leukotrienes, cytokines, and neuropeptides, all of which contribute to vasodilation, pruritus, and inflammatory cell recruitment. Therapies targeting these mediators provide adjunctive benefits but remain insufficient because they do not prevent mast cell activation. This limitation has prompted a paradigm shift toward upstream strategies designed to inhibit mast cell activation and degranulation. These strategies can be broadly divided into IgE-dependent approaches, targeting the cross-linking of the high-affinity IgE receptor, which include anti-IgE monoclonal antibodies, IgE traps, and Bruton’s tyrosine kinase inhibitors, and non-IgE-mediated approaches, such as cytokine blockade (interleukin [IL]-4, IL-5, IL-17, and IL-33), complement inhibition, c-kit targeting, Siglec-8 modulation, MRGPRX2 antagonism, and alarmin-directed therapies. Collectively, these advances signify a shift in chronic spontaneous urticaria (CSU) management from symptomatic mediator blockade to mechanism-based and personalized therapy. The ultimate objective is not merely to control symptoms but also to achieve true disease modification by altering the natural course of CSU.
INTRODUCTION
Chronic urticaria (CU) is defined as the spontaneous or inducible appearance of pruritic wheals, angioedema, or both lasting for > 6 weeks [1]. Its global prevalence is estimated at 0.5–1.4%, with chronic spontaneous urticaria (CSU) being the predominant subtype, occurring in the absence of identifiable external triggers [2,3]. CSU often coexists with inducible forms, which contribute to diagnostic and therapeutic challenges. Although not life-threatening, its recurrent and unpredictable course profoundly affects the quality of life, work productivity, and psychological well-being [2,4].
The natural course of CSU is highly variable, with disease duration ranging from several months to many years [5]. Epidemiological studies indicate that only 30–50% of patients achieve remission within 1–5 years, whereas many remain symptomatic for longer periods [5]. Prognostic indicators of prolonged disease have been suggested to include high disease activity, concomitant angioedema, comorbid autoimmune disorders, low total immunoglobulin E (IgE) levels, and elevated systemic inflammatory markers such as C-reactive protein (CRP) and D-dimer [6,7]. However, the strength and consistency of evidence for these biomarkers vary across studies, and their prognostic utility remains subject to ongoing debate [7]. Our longitudinal cohort study, using data-driven clustering, identified distinct subgroups of CU with heterogeneous remission and relapse trajectories, underscoring the variable natural course of the disease [8]. Patients with higher disease activity showed a prolonged disease course and poorer outcomes. In addition, female sex and IgE sensitization to house dust mites have been identified as independent predictors of unfavorable prognosis [8]. These observations are consistent with findings from an international real-world registry of CU, which reported that women, particularly those aged 30–65 years, tend to have more severe and refractory CSU, with greater disease burden and healthcare utilization [9]. These findings emphasize that the prognosis of CSU is influenced by disease activity and systemic inflammation, as well as immunological and demographic factors.
Despite the therapeutic advances, substantial unmet clinical needs persist. More than half of patients continue to experience symptoms despite standard doses of non-sedating H1-antihistamines (H1AHs), which are the guideline-recommended first-line therapy [10]. Even with up to four times the standard regimen, many patients remain uncontrolled [11]. These refractory cases emphasize the inherent limitations of antihistamines, which primarily block histamine H1 receptors but fail to target the broader immune dysregulation and diverse effector pathways underlying CSU.
This therapeutic gap has prompted the development of novel mechanism-based therapies targeting immune dysregulation, mast cell activation, autoimmunity, and cytokine signaling pathways. A comprehensive understanding of CSU pathogenesis and identification of precise therapeutic targets are essential to enhance disease control, achieve sustained remission, and ultimately modify the natural course of the disease through personalized treatment approaches.
PATHOPHYSIOLOGICAL INSIGHTS INTO CSU
Mast cells are the central effector cells in CSU that integrate immune, neuronal, and environmental signals that culminate in the development of itchy wheals and angioedema (Fig. 1). Upon activation, they release histamine and other mediators, including platelet-activating factor (PAF), leukotrienes, prostaglandins, and various cytokines [12]. These mediators disrupt endothelial integrity, promote vasodilation and plasma extravasation, stimulate sensory nerves to induce pruritus, and recruit inflammatory leukocytes such as eosinophils, neutrophils, basophils, and monocytes, amplifying cutaneous inflammation [6].
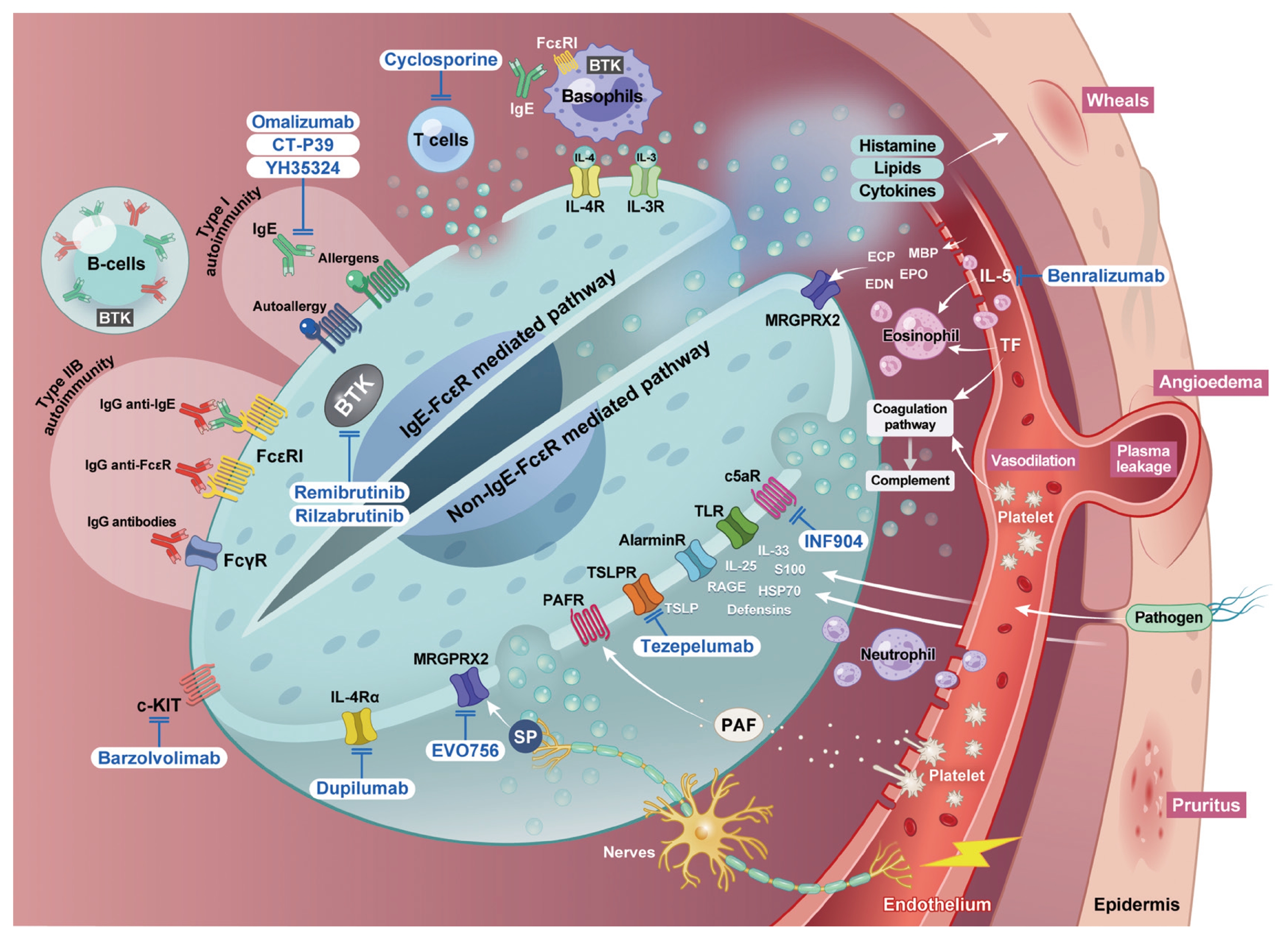
Pathophysiology and therapeutic targets in CSU. Mast cells function as the central effectors in CSU by integrating immunological, neuronal, vascular, and environmental signals. They can be activated via IgE-FcɛRI cross-linking (type I autoallergy) or IgG autoantibodies against FcɛRIα or IgE (type IIb autoimmunity). In addition, non-IgE pathways involving cytokine receptors (IL-4Rα, IL-33R, IL-17R, TSLPR), complement components (C3aR, C5aR), G protein–coupled receptors (PAFR, leukotriene receptors, MRGPRX2), and pattern recognition receptors (TLRs) converge to trigger mast cell activation. Activated mast cells release histamine, PAF, leukotrienes, prostaglandins, and cytokines, leading to endothelial damage, vasodilation, and plasma leakage. These mediators also stimulate sensory nerves, causing pruritus and recruiting eosinophils, neutrophils, basophils, and monocytes. The recruited effector cells, together with epithelial alarmins (IL-33, TSLP, and IL-25) and Th2/Th17 cytokines, amplify and sustain cutaneous inflammation. Major therapeutic targets include IgE/FcɛRI signaling (anti-IgE, IgE trap, BTK inhibitors), cytokine modulation (anti-IL-4, IL-5, and TSLP), c-kit blockade, complement inhibition, and emerging strategies such as MRGPRX2 antagonism or alarmin-directed therapy. CSU, chronic spontaneous urticaria; Ig, immunoglobulin; FcɛRI, high-affinity IgE receptor; FcγR, Fc gamma receptor; IL, interleukin; IL-4Rα, interleukin-4 receptor alpha; TSLP, thymic stromal lymphopoietin; TSLPR, thymic stromal lymphopoietin receptor; C3aR, complement component 3a receptor; C5aR, complement component 5a receptor; PAF, platelet-activating factor; PAFR, platelet-activating factor receptor; MRGPRX2, Mas-related G protein–coupled receptor member X2; TLR, toll-like receptor; Th, T helper cell; BTK, Bruton’s tyrosine kinase; ECP, eosinophil cationic protein; EDN, eosinophil-derived neurotoxin; EPO, eosinophil peroxidase; MBP, major basic protein; RAGE, receptor for advanced end-products; S100, S100 calcium-binding protein family; SP, substance P; HSP70, heat shock protein 70; TF, tissue factor.
Mast cell activation in CSU occurs through multiple mechanisms. The best-characterized pathway involves IgE-dependent cross-linking of the high-affinity IgE receptor (FcɛRI), which triggers Lyn and Syk kinase activation, Bruton’s tyrosine kinase (BTK) signaling, calcium influx, and rapid degranulation [12,13]. Autoimmune mechanisms provide additional routes of activation (Table 1). Type I (autoallergic) CSU is mediated by autoreactive IgE antibodies directed against self-antigens such as thyroid peroxidase, double-stranded DNA, or interleukin (IL)-24 [14,15]. These autoreactive IgEs cross-link FcɛRI on mast cells and basophils, inducing degranulation in the absence of external allergens. Patients with this endotype typically exhibit atopic features, elevated total IgE levels, and a favorable response to anti-IgE therapy [16]. In contrast, type IIb autoimmune CSU is driven by IgG autoantibodies targeting FcɛRIα, IgE, or thyroid peroxidase [17–19]. These antibodies can directly activate mast cells and basophils, often through complement involvement. Our group further demonstrated that anti-heat shock protein (HSP)10 IgG autoantibodies are elevated in CSU and correlate with disease activity and PAF-dependent mast cell degranulation [20]. Clinically, this endotype is associated with more severe disease, a poor response to H1AHs and omalizumab, lower total IgE levels, and positive autologous serum skin test (ASST) or basophil activation test (BAT). These autoimmune pathways collectively account for the heterogeneity of CSU and the variability in biomarker profiles and treatment responses.
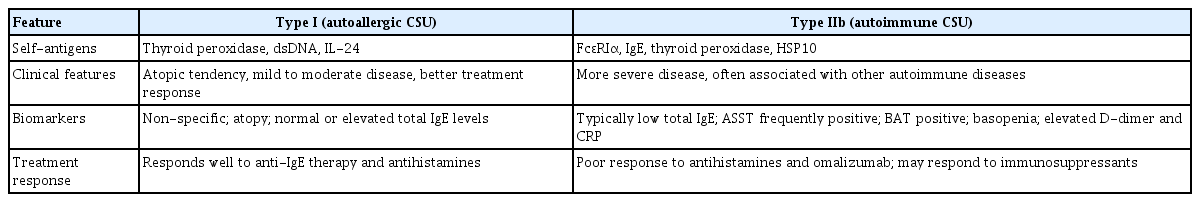
Two autoimmune endotypes of CSU
Mast cells integrate diverse external stimuli through an extensive receptor repertoire. Cytokine receptors such as IL-4Rα, IL-33R (ST2), IL-17R, and the thymic stromal lymphopoietin (TSLP) receptor enable them to detect type 2 inflammatory signals and epithelial alarmins [21]. Complement receptors (C3aR and C5aR) mediate responses to anaphylatoxins [22]. Among G protein-coupled receptors, leukotriene, purinergic, and PAF receptors play pivotal roles, whereas the mas-related G protein-coupled receptor member X2 (MRGPRX2) has emerged as a key mediator of IgE-independent mast cell activation. MRGPRX2 is triggered by neuropeptides, antimicrobial peptides, and several clinically relevant drugs, including fluoroquinolones, vancomycin, neuromuscular blocking agents, and radiocontrast media [23,24]. In addition, pattern-recognition receptors, such as toll-like receptors and NOD-like receptors, connect mast cells to innate immune sensing and host defense mechanisms [25,26].
Activated mast cells orchestrate a broad immune network [6, 12,14, 27]. Type 2 cytokines—particularly IL-4, IL-5, and IL-13—promote IgE synthesis, eosinophil recruitment, and upregulation of FcɛRI expression [14]. IL-17A, which is derived from Th17 cells and innate immune sources, enhances mast cell activation and neutrophil infiltration [28]. Impaired regulatory T-cell function further permits autoreactive immune responses. Epithelial alarmins, including IL-33, TSLP, and IL-25, prime mast cells, basophils, and group 2 innate lymphoid cells to reinforce type 2 inflammation [29]. The complement fragments C3a and C5a further potentiate mast cell degranulation [22]. Eosinophils and neutrophils recruited to the lesional skin release cytotoxic proteins and proinflammatory cytokines, contributing to vascular leakage and tissue injury [30]. Basophils function as key effector cells in CSU, exhibiting increased expression of the activation marker CD203c compared to healthy controls, whereas their circulating numbers are often reduced in active disease, correlating with disease severity [8, 31,32]. Basophils release histamine, leukotrienes, and Th2 cytokines such as IL-4 and IL-13, which enhance IgE production and sustain mast cell activation. Autoreactive B cells generate IgE and IgG autoantibodies, including anti-FcɛRIα, anti-IgE, and anti-HSP10, whereas Th2 and Th17 subsets amplify mast cell and eosinophil activation [15, 18, 20, 28, 33]. Impaired T- and B-regulatory cell function perpetuates autoreactivity and chronic inflammation [15, 34, 35].
In addition to immune cells, other tissue-resident and circulating effector systems contribute to the pathogenesis of CSU. Endothelial cells activated by histamine, PAF, and vascular endothelial growth factor, disrupt vascular barrier integrity and promote plasma leakage and leukocyte trafficking [36]. Afferent sensory nerves respond to histamine, substance P, and proteases via pruritic signaling and neurogenic inflammation, whereas neuropeptides activate mast cells through MRGPRX2 [24]. Platelets are increasingly recognized as immune modulators that interact with mast cells via PAF and serotonin release, and heightened platelet activation has been associated with CSU severity and thromboinflammatory responses [36–38].
Taken together, CSU represents a mast cell–driven yet immunologically complex disorder in which IgE, autoantibodies, cytokines, alarmins, neuropeptides, complement, and multiple immune and vascular elements interact to sustain chronic inflammation. This multifaceted pathogenesis emphasizes the need for therapies that extend beyond H1AHs and anti-IgE agents and target disease mechanisms to enable personalized and more effective management strategies.
BEYOND ANTIHISTAMINES: MECHANISM-ORIENTED THERAPEUTIC STRATEGIES
Effector mediator-targeted therapies
Histamine is the principal mediator of CSU symptoms, and non-sedating H1AHs remain the recommended first-line therapy. However, up to half of patients remain symptomatic even after a four-fold dose escalation [1]. In addition to histamine, mast cells release additional mediators, including PAF, leukotrienes, prostaglandins, and cytokines [21].
Leukotriene receptor antagonists (LTRAs) such as montelukast have been explored as adjunctive therapy, particularly in patients with aspirin- or NSAID-exacerbated urticaria [39]. However, international guidelines discourage their routine use because of limited and low-quality supporting evidence [40]. A recent meta-analysis showed that adding an LTRA to H1AH therapy modestly reduced urticaria severity and improved quality of life without increasing adverse events [41].
PAF has emerged as a key mediator, particularly in antihistamine-refractory CSU [36]. Our previous study demonstrated significantly higher serum PAF levels and lower PAF-acetylhydrolase activity in patients with CSU than in healthy controls, with elevated PAF concentrations noted among H1AH non-responders, supporting its potential as a therapeutic target [42]. Currently, there are no selective PAF antagonists available. Rupatadine, a dual H1AH and PAF receptor antagonist, has been shown to significantly reduce pruritus and improve quality of life in randomized trials; however, the overall therapeutic benefit remains modest [43,44]. Other lipid mediator–targeting strategies, including thromboxane modulation and CRTH2 antagonism, have been evaluated in small studies but lack consistent evidence to support their clinical use [45,46].
IgE-FcɛRI-mediated therapies: anti-IgE and BTK inhibitors
The IgE–FcɛRI signaling represents a central mechanism in the pathogenesis of CSU, driving mast cell and basophil activation and subsequent mediator release [14, 47–49]. In addition to its direct effects on mast cells, IgE depletion exerts broader immunomodulatory actions [50–55]. Downregulation of the low-affinity IgE receptor FcɛRII (CD23) on B cells and dendritic cells indirectly suppresses IgE synthesis and IgE-dependent antigen presentation, while anti-IgE therapy has been associated with reductions in circulating eosinophil counts [47,48, 50–54].
Therapeutic strategies targeting this axis can broadly be di- vided into upstream and downstream approaches (Table 2). Upstream inhibition aims to prevent IgE binding and cross- linking of FcɛRI through neutralization of free IgE using monoclonal antibodies or engineered IgE traps, thereby reducing FcɛRI occupancy and inducing gradual downregulation of FcɛRI expression on effector cells [47–55]. In parallel, downstream blockade of FcɛRI signaling represents an alternative strategy to suppress mast cell activation. Inhibition of BTK, a key mediator of FcɛRI signal transduction, can attenuate mast cell activation, including activation driven by IgG autoantibodies [56].
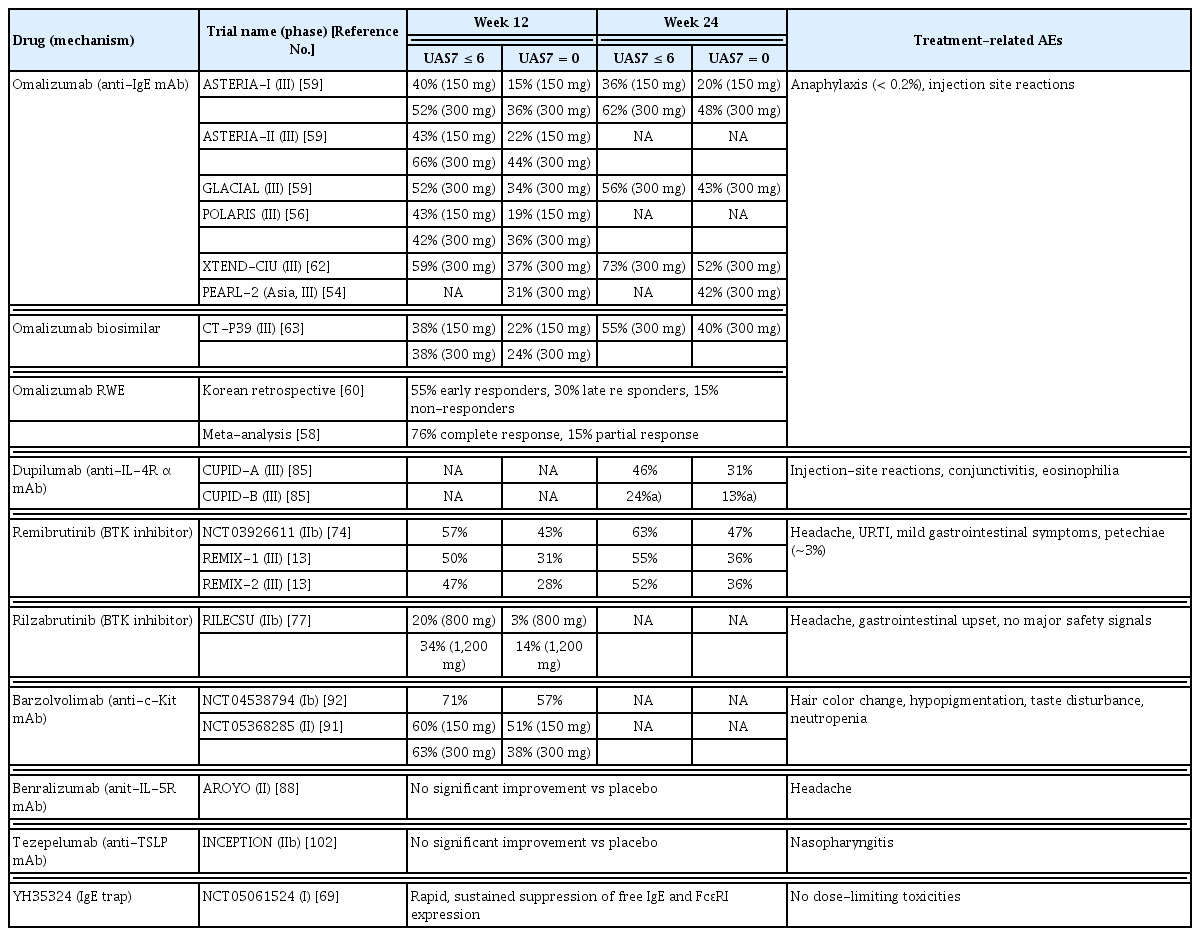
Summary of key clinical trials of therapeutics based on UAS7 scores in chronic spontaneous urticaria
Anti-IgE therapies
Omalizumab is a well-established second-line treatment for antihistamine-refractory CSU [1, 10, 53]. Large randomized controlled trials (ASTERIA I/II, GLACIAL) and multiple real-world studies have consistently demonstrated its efficacy, with significant reductions in UAS7 scores, improvements in CU-specific quality of life, and rapid symptom relief, often within the first 1–2 weeks of therapy [55, 57-61]. Its long-term safety profile remains favorable, with anaphylaxis being rare (< 0.2%) and > 95% of cases occurring within the first 3 months of treatment [59]. Its efficacy and safety have been validated in diverse populations, including children, pregnant women, and older adults [60]. Current guidelines recommend the subcutaneous administration of 150–300 mg every 4 weeks [10]. Approximately 60% of patients achieve well-controlled disease within 6 months, and 20–40% may attain complete remission, depending on the study population and response criteria [60-64].
Our retrospective longitudinal cohort of 386 patients treated with omalizumab for > 6 months demonstrated progressively increasing cumulative response rates of 55.2% at 3 months, 71.0% at 6 months, and 81.4% at 9 months [62]. To improve treatment accessibility, an omalizumab biosimilar was approved in 2025. In a phase III trial (NCT0442689), the omalizumab biosimilar CT-P39 exhibited therapeutic equivalence to the reference omalizumab 300 mg in terms of change in weekly itch severity score (difference, 0.77; 95% confidence interval, −0.37 to 1.90), with parallel reductions in UAS7 and sustained efficacy after switching [65]. Its introduction is expected to enhance cost-effectiveness and broaden patient access; however, confirmation from long-term real-world data is warranted. Nevertheless, approximately 22% of patients exhibit a persistently poor response from as early as 3 months onward, underscoring the need for biomarkers to identify non-responders [62]. A low baseline total IgE level (< 40 kU/L) has consistently been identified as a predictor of a poor response to omalizumab. The clinical and laboratory features associated with non-response often overlap with the characteristics of type IIb autoimmune CSU, including basopenia, autoreactivity (positive basophil activation or ASSTs), elevated systemic inflammatory markers, and concomitant autoimmune diseases [6, 62, 66-69]. These findings emphasize the importance of endotype-driven therapeutic strategies in guiding patient selection for omalizumab treatment.
Ligelizumab, a humanized anti-IgE monoclonal antibody with higher IgE-binding affinity than omalizumab, has been evaluated for CSU [54,55]. It more effectively suppresses free IgE levels and downregulates FcɛRI expression in preclinical models and early-phase clinical studies [54]. In phase III clinical trials, ligelizumab demonstrated numerically greater reductions in urticaria activity scores than omalizumab; however, it did not consistently achieve superiority across the primary clinical endpoints [55]. Consequently, its clinical development for CSU was discontinued, and a next-generation approach to IgE neutralization involves the development of IgE traps and engineered fusion proteins designed to bind IgE with a markedly higher affinity [70,71]. YH35324, the first-in-class IgE trap, is a long-acting IgE Trap-Tc fusion protein comprising the extracellular domain of human FcɛRIα fused to a human IgD/IgG4-modified Fc region. In a phase 1 clinical trial, YH35324 was well tolerated, with no dose-limiting toxicities, and demonstrated a favorable pharmacokinetic and safety profile. Treatment led to rapid and sustained suppression of free IgE levels and FcɛRI expression [71,72]. Although the efficacy data for CSU are not yet available, the IgE trap strategy offers potential advantages over conventional anti-IgE therapy by enabling more complete IgE depletion and broader downregulation of IgE-mediated pathways.
BTK inhibitors
Beyond targeting IgE directly, inhibition of downstream FcɛRI signaling has emerged as a promising therapeutic strategy. This approach is particularly relevant in type IIb autoimmune CSU, where anti-FcɛRI or anti-IgE autoantibodies drive mast cell activation independent of free IgE. In such cases, blocking FcɛRI-mediated signaling through BTK inhibition may help overcome resistance to anti-IgE therapies [56].
Fenebrutinib, a first-generation oral BTK inhibitor, has demonstrated efficacy in a phase II trial for H1AH-refractory CSU [73]. Preclinical studies further indicated that BTK blockade may reduce the production of anti-FcɛRIα autoantibodies, implicating BTK in autoreactive B-cell function in type IIb autoimmune CSU [74]. However, clinical development of fenebrutinib was discontinued because of its hepatotoxicity.
Since then, more selective BTK inhibitors have been developed, and remibrutinib has emerged as the most advanced candidate [75]. The phase III REMIX-1 and REMIX-2 trials demonstrated rapid and significant reductions in urticaria activity compared to placebo. At week 12, approximately 48% of remibrutinib-treated patients achieved well-controlled disease (UAS7 ≤ 6), and 29% attained complete control (UAS7 = 0) [13]. Patients who switched from placebo to remibrutinib after week 12 experienced rapid improvements that were sustained through week 24. Notably, the efficacy was consistent regardless of prior omalizumab exposure [59].
Remibrutinib was generally well-tolerated, with an overall adverse event profile comparable to placebo. A characteristic treatment-related event was petechiae, reflecting BTK’s role in platelet signaling, which is typically mild, transient, and occurs slightly more frequently (approximately 3%) than in the placebo group [13]. Although BTK is essential in B-cell receptor signaling and could theoretically impair immunoglobulin synthesis or increase infection risk, recent studies have demonstrated no clinically meaningful effects on IgG, IgA, or IgM levels, and the infection rates were comparable to placebo [76]. Despite its promising efficacy and safety profile, remibrutinib has not yet received FDA approval, and long-term safety data remain limited. Real-world studies across diverse geographic and ethnic populations are crucial to confirm its sustained efficacy, safety, and overall risk–benefit profile.
Rilzabrutinib, another next-generation oral BTK inhibitor with enhanced selectivity and prolonged BTK occupancy, has demonstrated dose-dependent suppression of FcɛRI-mediated basophil and mast cell activation in phase I/II studies, accompanied by reductions in serum biomarkers, such as IL-31, soluble MRGPRX2, and autoantibodies (anti-TPO IgG and anti-FcɛRIα IgG) [77,78]. In a phase II trial, rilzabrutinib at a dose of 1200 mg/day significantly improved UAS7 scores compared with placebo at week 12 [78]. Ongoing phase III trials are expected to elucidate the clinical efficacy and long-term safety of H1AH-refractory CSU. Rilzabrutinib has thus far exhibited a favorable safety profile, with most adverse events being mild and transient, including headache and gastrointestinal symptoms.
Conventional immunomodulators
Cyclosporine, a calcineurin inhibitor, has long been used as a steroid-sparing immunomodulator for atopic dermatitis and psoriasis, and has also been investigated in H1AH-refractory CSU [1]. Its mechanism involves the inhibition of T-cell receptor signaling and IL-2 transcription through calcineurin blockade [79]. In CSU, cyclosporine appears to exert additional effects by suppressing histamine release from mast cells and basophils, and modulating autoreactive immune responses [80,81].
Evidence from small randomized controlled trials and meta-analyses supports its efficacy in CSU, demonstrating significant improvements in UAS7 scores and quality of life compared with placebo. A recent systematic review reported response rates of approximately 54% at 4 weeks and 73% at 12 weeks, even with doses ≤ 3 mg/kg/day, supporting the effectiveness of low-dose regimens [82,83]. Moreover, cyclosporine appears to be particularly beneficial for patients with type IIb autoimmune CSU, who often respond poorly to omalizumab [6, 83,84]. Among these endotypes, cyclosporine is a valuable alternative therapeutic option when omalizumab is unavailable or ineffective.
However, cyclosporine use is limited by its safety profile. Adverse effects include hypertension, nephrotoxicity, dyslipidemia, and gastrointestinal symptoms, with the risks increasing with dose and treatment duration [10, 82]. Therefore, international guidelines recommend cyclosporine as a third-line therapy for selected severe cases [1]. Careful monitoring is essential; blood pressure and renal function should be assessed regularly (every 2–3 mo), and treatment should be limited to the shortest effective duration to minimize toxicity. With the advent of biologics, the role of cyclosporine is diminishing but remains valuable in selected patients, particularly in resource-limited settings.
Non-IgE-mediated therapeutic strategies
Cytokine blockade
Cytokines such as IL-4, IL-5, IL-13, IL-17, and IL-33 are important mediators of CSU pathogenesis [32,33, 85]. Dupilumab, an IL-4Rα antagonist that blocks both IL-4 and IL-13 signaling, has demonstrated clinically meaningful efficacy in H1AH-refractory CSU. In the phase III LIBERTY-CSU CUPID trials, dupilumab significantly improved UAS7 scores and quality of life compared with placebo [86]. In omalizumab-naïve patients, up to 30% achieved a complete response at week 24 and approximately 60% achieved well-controlled disease [86]. Conversely, in omalizumab non-responders, the efficacy was less pronounced, emphasizing the importance of appropriate patient selection. Based on these results, dupilumab has been approved for CSU in Japan, the UAE, and the USA in patients aged ≥ 12 years with a disease uncontrolled by H1AHs. Dupilumab exhibits a favorable safety profile, with the most common adverse events being injection-site reactions, conjunctivitis, and transient eosinophilia. Notably, no increase in serious adverse events or anaphylaxis was observed compared to the placebo, and long-term data from other indications, such as atopic dermatitis and asthma, support its continued safety.
Other cytokine-targeted strategies remain investigational. IL-5 blockade and IL-17 inhibition have shown anecdotal or preliminary benefits in small case series, but the evidence remains inconsistent [87,88]. Notably, a recent phase IIb trial of benralizumab failed to demonstrate its clinical efficacy despite complete eosinophil depletion, implying a limited role for eosinophils in CSU [89]. Anti-TSLP therapy with tezepelumab was recently evaluated in the phase IIb INCEPTION trial in H1AH–refractory CSU; however, our findings did not show a clinically meaningful benefit over placebo, limiting enthusiasm for further development in CSU [90].
Mast cell depletion
An alternative therapeutic approach for CSU is direct depletion of mast cells, which are the central effector cells in disease pathogenesis. Unlike strategies that inhibit downstream mediators or upstream IgE pathways, mast cell depletion eliminates primary sources of histamine, lipid mediators, and cytokines by modulating mast cell survival and proliferation.
Barzolvolimab, a humanized monoclonal antibody targeting the receptor tyrosine kinase c-kit (CD117), induces apoptosis and sustained depletion of dermal mast cells, accompanied by a marked reduction in serum tryptase [91]. In a phase II trial, barzolvolimab 150 mg every 4 weeks and 300 mg every 8 weeks significantly improved the UAS7 and UCT scores at week 12 compared to the placebo, with up to 71% of patients achieving a complete response in the 52-week analysis [92,93]. Barzolvolimab was generally well tolerated. However, unique on-target adverse effects, including hair color changes, skin hypopigmentation, taste disturbances, and transient neutropenia, have been reported, warranting careful monitoring in ongoing phase III trials [93]. Because mast cells contribute to host defense, tissue repair, and immune regulation, the long-term consequences of sustained mast cell depletion require further evaluation in larger and longer studies.
MRGPRX2 antagonists
MRGPRX2 has emerged as a potential therapeutic target for CSU. Skin biopsies from affected patients, particularly those with severe or H1AH–refractory disease, show increased MRGPRX2 expression in lesional mast cells compared to healthy controls, and elevated serum levels have been correlated with disease activity [94,95]. Moreover, recent studies indicate that IgE can upregulate MRGPRX2 expression in mast cells, enhancing their responsiveness to neuropeptides, antimicrobial peptides, and certain drugs [96–98]. This cross-talk between the IgE-FcɛRI and MRGPRX2 pathways may explain why patients with high total or autoreactive IgE levels exhibit exaggerated mast cell activation even in the absence of allergen exposure.
Based on these findings, the pharmacological inhibition of MRGPRX2 has been proposed as a novel therapeutic approach. EVO756, a first-in-class oral small-molecule antagonist, has shown potent dose-dependent inhibition of MRGPRX2-mediated mast cell degranulation in preclinical studies while sparing IgE-dependent signaling [99,100]. Early phase clinical trials are ongoing, although the efficacy data for CSU are not yet available.
Although experimental, MRGPRX2 antagonists may be particularly relevant for CSU subgroups characterized by neurogenic inflammation, drug hypersensitivity, or non-IgE-mediated mast cell activation. Future studies should focus on identifying biomarkers of MRGPRX2-driven disease and establishing the efficacy and long-term safety of these agents in large-scale CSU populations.
Emerging targets and experimental approaches
Complement-derived anaphylatoxins, particularly C5a, can directly activate mast cells and basophils, implicating the role of the complement system in autoimmune CSU [101]. Although agents such as C1s and C5a inhibitors are being evaluated in other autoimmune diseases, including avacopan for ANCA-associated vasculitis, their clinical efficacy in CSU has yet to be established [102,103].
Sialic acid-binding immunoglobulin-like lectin 8 (Siglec-8), which is selectively expressed on mast cells and eosinophils, represents a promising target for mast cell silencing and eosinophil depletion [104,105]. Lirentelimab, a humanized Siglec-8 monoclonal antibody, demonstrated clinical activity in early phase trials [106]; however, subsequent phase III studies on eosinophilic gastrointestinal diseases failed to meet their primary endpoints, and further development in CSU has not been pursued.
Alarmins such as IL-25, IL-33, and TSLP are released upon tissue injury or stress and normally promote tissue repair and restoration of homeostasis [29, 85]. In CSU, sustained elevation of alarmins may perpetuate mast cell activation and type 2 inflammation rather than resolve it. Moreover, damage-associated molecules, including HSPs and haptoglobin, have been implicated in CSU pathogenesis; autoantibodies against these alarmins, such as anti-HSP10 IgG, may disrupt their regulatory functions, impair inflammation resolution, and sustain chronic diseases [20].
Epigenetic dysregulation is another layer of complexity. Altered miRNA profiles in CSU have been associated with disease activity, treatment refractoriness, and the presence of anti-HSP10 autoantibodies, indicating that miRNAs may modulate alarmin pathways, mast cell activation thresholds, and autoimmune responses [20, 107,108]. From a translational perspective, miRNA signatures hold promise as endotype-defining biomarkers; however, therapeutic modulation using miRNA inhibitors or mimics, already explored in other immune-mediated disorders, may offer a future strategy for disease modification in CSU [109].
TOWARD PERSONALIZED AND PRACTICAL MANAGEMENT IN CSU
The recognition of CSU as a heterogeneous immune-mediated disorder emphasizes the importance of endotype-oriented management. Biomarkers, such as baseline total IgE, basophil CD203c expression, ASST, BAT, and autoreactive IgG or IgE autoantibodies, can help differentiate type I autoallergy from type IIb autoimmune CSU. Low total IgE levels (< 40 kU/L), basopenia, and elevated levels of systemic inflammatory markers (CRP and D-dimer) consistently correlate with poor omalizumab response and more severe disease, supporting their value in treatment stratification [12, 32, 62, 66]. Emerging biomarkers, including serum cytokine profiles, proteomic patterns, and dysregulated miRNAs, may further refine endotyping and advance precision medicine for CSU.
However, translating these mechanistic insights into clinical practice remains challenging. Although current guidelines recommend a stepwise approach using H1AHs, omalizumab, cyclosporine, or emerging biologics, real-world management is often complex and individualized. Moreover, outcomes observed in clinical trials may not fully reflect those observed during routine care. Even with newer agents such as BTK inhibitors or dupilumab, the proportion of complete responders remains comparable to that of omalizumab, emphasizing the need for real-world data and predictive biomarkers to optimize therapy selection according to patient endotypes [13, 91]. For instance, BTK inhibitors may be particularly effective in type IIb autoimmune CSU, whereas dupilumab may be more suitable for patients with type 2-dominant inflammation. Integrating biomarker-based panels into therapeutic algorithms could facilitate the early identification of non-responders and reduce treatment delays.
Practical considerations also influence long-term management. Biologics and targeted therapies are expensive, raising concerns about their cost-effectiveness, accessibility, and optimal treatment duration. Strategies for dose tapering, discontinuation, and prediction of sustained remission remain poorly defined. Additionally, regional variations in drug approval and reimbursement contribute to disparities in care. Future studies should focus on validating biomarkers for routine clinical use, linking them to therapeutic algorithms, and generating robust real-world evidence across diverse populations to achieve personalized and pragmatic CSU management.
CONCLUSION AND PERSPECTIVES
CSU is a mast-cell–driven yet immunologically complex disease involving IgE, autoantibodies, cytokines, alarmins, complement, and epigenetic regulation. This multifaceted pathophysiology underlies the heterogeneity of the disease course and therapeutic responses, emphasizing the need to move beyond symptomatic mediator blockade.
Recent therapeutic advances, including anti-IgE agents, BTK inhibitors, cytokine blockade, mast cell depletion, and other novel approaches, represent a transition toward mechanism-based, endotype-driven treatments. However, a substantial proportion of patients continue to experience incomplete remission and the gap between clinical trial outcomes and real-world effectiveness persists. Validated biomarkers are required to guide individualized therapy, predict prognosis, and identify patients at risk for treatment refractoriness.
Ultimately, CSU management should extend beyond symptom control to true disease modification to restore immune tolerance and prevent chronicity. Achieving this goal requires the integration of biomarker-driven precision medicine, longitudinal real-world evidence, and emerging targeted therapies, thus paving the way for more effective and personalized care for patients with CSU.
Notes
Acknowledgments
The author sincerely thanks Ms. Woo Hyun Cho from the Public Relations Office of Ajou University Hospital for her technical assistance in preparing the illustration for Figure 1.
Conflicts of interest
Ye YM has served as a consultant, speaker, and research investigator for Novartis, Yuhan, Amgen, and Celldex.
Funding
This work was supported by a grant from the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT and Future Planning (MSIP) of Korea (2022R1A2C2006607) and the GRRC program of Gyeonggi province (GRRCAjou2023-B02).