Germline DDX41 mutations in myeloid neoplasms: a comprehensive review
Article information

Abstract
DEAD-box helicase 41 (DDX41)-associated myeloid neoplasms represent a distinct hereditary cancer syndrome and have emerged as the most common form of genetic predisposition to adult-onset hematologic malignancies, accounting for 3–5% of adults with myelodysplastic syndrome and acute myeloid leukemia. DDX41 is a multifunctional DEAD-box RNA helicase that plays essential roles in pre-mRNA splicing, ribosome biogenesis, and innate immune regulation. Pathogenic germline variants predominantly cluster within the N-terminal region, whereas somatic second-hit mutations are concentrated in the C-terminal helicase domain. The characteristic “two-hit” model involves a germline loss-of-function mutation followed by a somatic variant—most commonly p.R525H—leading to malignant transformation. This process occurs through defective small nucleolar RNA processing, impaired ribosome assembly, erythropoietic abnormalities, and R-loop–mediated DNA damage. Clinically, the phenotype is consistent and characterized by late-onset disease in the sixth to seventh decades of life, a strong male predominance, hypocellular bone marrow, and normal cytogenetics. Despite these distinctive features, affected individuals generally exhibit favorable treatment responses and outcomes compared with sporadic cases. An important clinical consideration in DDX41-associated disease management is the planning of allogeneic hematopoietic cell transplantation, where genetic screening for DDX41 variants among potential related donors is essential. Future investigations should aim to elucidate the molecular mechanisms underlying DDX41-driven leukemogenesis, refine surveillance strategies, and develop targeted therapeutic approaches.
INTRODUCTION
The understanding of genetic predisposition to cancer has evolved remarkably over the past century. What began as clinical observations of familial cancer clustering has transformed into the precise identification of specific germline pathogenic variants, facilitated by major advances in molecular biology and next-generation sequencing (NGS) technologies [1–4]. Hereditary cancer syndromes account for approximately 5–10% of all cancer cases and up to 17.5% of advanced cancer cases [5]. This paradigm shift has fundamentally reshaped approaches to cancer risk assessment and prevention.
Myeloid malignancies with genetic predisposition were once considered rare. However, recognition of familial clustering in these malignancies has progressed from anecdotal reports in the mid-20th century to a sophisticated molecular understanding of hereditary predisposition syndromes [6–9]. In parallel with this progress, germline deleterious variants in multiple genes are now known to predispose individuals to hematopoietic malignancies, and recent leukemia classification systems have been revised to include these entities [10–12]. For example, the fifth edition of the World Health Organization Classification of Haematolymphoid Tumors categorizes myeloid neoplasms with germline predisposition into 18 distinct syndromes based on disease phenotype and causative genes [12]. Current evidence indicates that about 10% of individuals with myeloid malignancies carry pathogenic germline variants in established predisposition genes [13], with DEAD-box helicase 41 (DDX41) mutations representing the most prevalent subset in adults. Distinct predisposition syndromes display unique epidemiological patterns: classical hereditary bone marrow failure syndromes and RUNX1-related disorders often present earlier with syndromic features, whereas DDX41-associated disease typically manifests in adulthood [13,14].
DDX41 is the most common germline predisposition gene implicated in adult-onset myeloid neoplasms, identified in approximately 3–5% of adults with myelodysplastic syndrome (MDS) and acute myeloid leukemia (AML) [13,15]. About 40–50% of myeloid neoplasms harboring germline DDX41 variants acquire an additional somatic mutation on the wild-type DDX41 allele [16–19]. More than 95% of these somatic mutations are missense, with p.R525H being the most frequent, accounting for approximately 60–80% of all somatic DDX41 variants [20]. DDX41 plays an essential role in hematopoiesis and performs diverse functions, including nucleic acid sensing [21–23] and nuclear RNA regulatory processes such as pre-mRNA splicing, R-loop resolution, and ribosome biogenesis [20,24–26]. Experimental models suggest that impaired R-loop resolution—resulting in innate immune activation [27–29] and defective ribosome biogenesis [30–32] contribute to the pathogenesis of myeloid neoplasms associated with germline DDX41 variants. However, the precise mechanisms by which pathogenic germline DDX41 mutations lead to malignant transformation remain unclear. This review summarizes current knowledge on DDX41’s molecular functions, germline variant spectrum, pathogenic mechanisms, clinical phenotype, treatment outcomes, prognosis, and diagnostic and management considerations.
STRUCTURE AND FUNCTIONS OF DDX41
The DDX41 gene is located on chromosome 5q35.3, a region frequently implicated in myeloid neoplasms. DDX41 exhibits the canonical structure of DEAD-box RNA helicases, consisting of several distinct domains that enable nucleic acid recognition, adenosine triphosphate (ATP) hydrolysis, and RNA remodeling. The protein contains an N-terminal segment that functions as a nuclear localization signal, followed by two highly conserved domains—the DEAD domain and the helicase domain—that form the catalytic core, as well as a C-terminal extension. The core structure of DEAD-box proteins comprises two primary RecA-like domains. The first RecA-like domain contains the characteristic DEAD motif together with Walker A and Walker B sequences essential for ATP binding and hydrolysis, whereas the second domain contains helicase motifs that mediate RNA binding and conformational rearrangements [24, 33].
DDX41 plays critical roles in several aspects of RNA metabolism, particularly in the regulation of pre-mRNA splicing and ribosome biogenesis. RNA helicases, in general, alter RNA structures and RNA–protein interactions through ATP-dependent helicase activity. DDX41 is thought to be integrated into the C complex of the spliceosome [28], which represents a central component in the two-step transesterification reactions that mediate RNA splicing [34]. Although the precise role of DDX41 in splicing remains incompletely defined, experimental studies have shown that its loss or mutation results in splicing alterations such as exon skipping and intron retention, especially in hematopoietic progenitor cells [19, 35]. DDX41 also contributes to the processing of pre-ribosomal RNA (rRNA), a critical step in ribosome assembly [31]. Small nucleolar RNAs (snoRNAs)—a class of noncoding RNAs—are essential for rRNA modification through 2′-O-methylation and pseudouridylation. Mutations in DDX41 have been linked to defective snoRNA processing, which subsequently impairs ribosome function [30]. This evidence highlights a novel role for DDX41 in connecting RNA splicing with ribosome biogenesis, as snoRNA processing is functionally intertwined with splicing regulation.
DDX41 also functions as a sensor of nucleic acids released from intracellular pathogens, including double-stranded DNA and cyclic di-guanosine monophosphate [21]. Under physiological conditions, DDX41 is primarily localized in the nucleus due to its N-terminal nuclear targeting sequence, where it participates in RNA splicing and ribosome biogenesis. Upon cellular activation or nucleic acid stimulation, however, DDX41 can translocate to the cytoplasm, where it acts as a pattern recognition receptor that detects foreign DNA and cyclic dinucleotides [24, 33]. DDX41 interacts with the adaptor protein stimulator of interferon genes (STING), initiating a STING-dependent innate immune response [23, 36]. This interaction is regulated by phosphorylation of DDX41 at tyrosine residue 414 by Bruton’s tyrosine kinase, which enhances the DDX41–STING association [37]. Nevertheless, whether this function contributes to hematologic malignancies remains unclear. Although germline DDX41 variants are generally loss-of-function and may reduce immune responsiveness, some studies suggest that DDX41 deficiency can paradoxically induce inflammation through R-loop accumulation [29].
In addition, DDX41 is involved in the regulation of R-loop formation, which consists of structures composed of DNA–RNA hybrids and displaced single-stranded DNA [38]. Excessive accumulation of R-loops is associated with pathological conditions, impaired transcriptional elongation, and genomic instability [28]. It has been proposed that DDX41 directly resolves DNA–RNA hybrids, thereby preventing activation of DNA damage signaling [26,29]. Loss-of-function variants of DDX41 lead to increased R-loop accumulation and enhanced inflammatory signaling through the STING–TBK1 axis [39]. Although DDX41 exhibits annealing activity, its helicase unwinding function is crucial for resolving R-loops. Mutations such as p.R525H impair this activity, potentially promoting activation of the STING–TBK1 pathway [36].
In summary, DDX41 is a multifunctional DEAD-box RNA helicase with essential roles in RNA splicing, ribosome biogenesis, innate immune regulation, and R-loop resolution (Fig. 1). Mutations in DDX41 are linked to myeloid malignancies by disrupting these fundamental cellular processes and promoting disease pathogenesis.
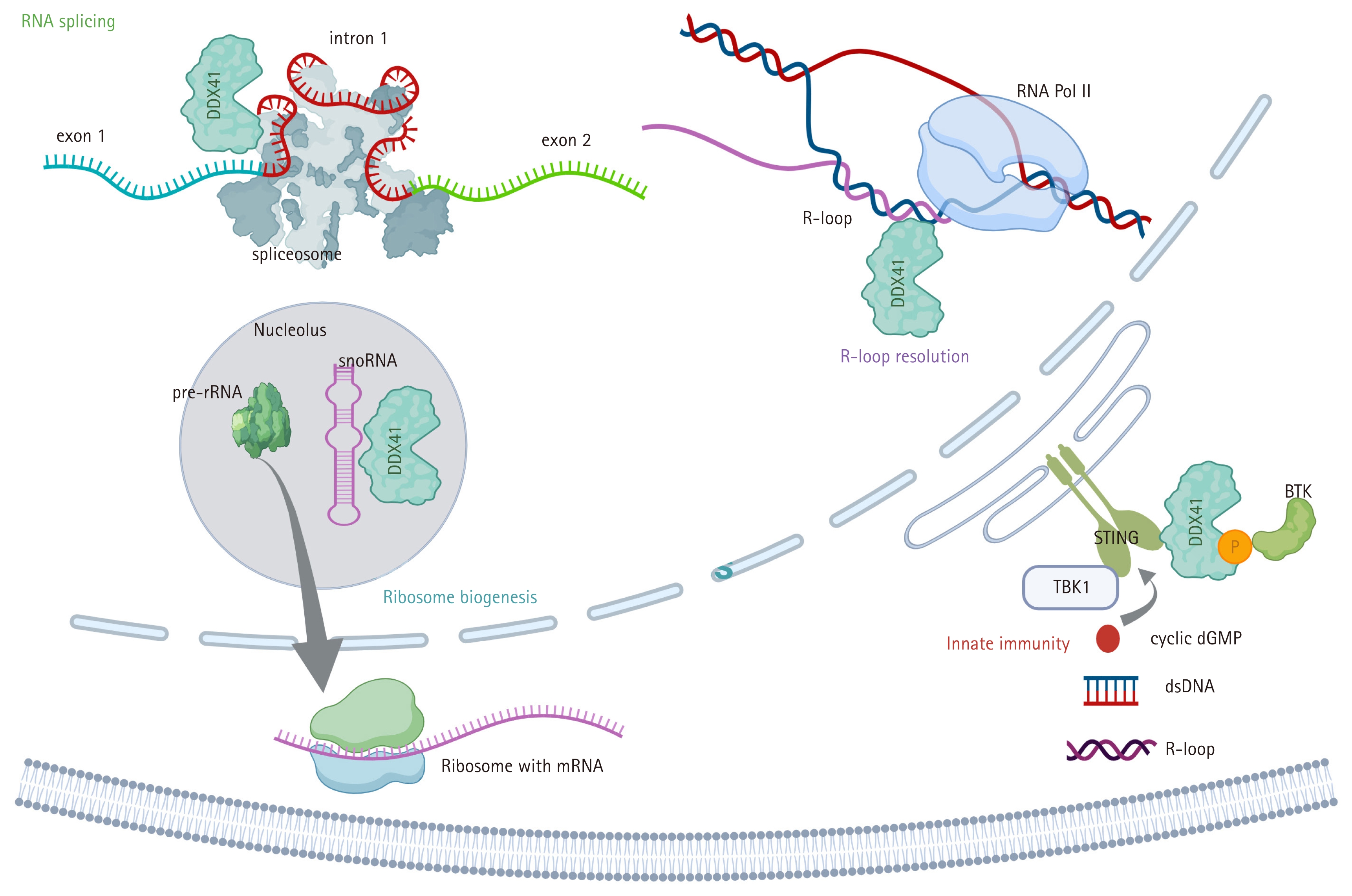
Molecular functions of DDX41 across cellular compartments. Depicted functions include RNA splicing, ribosome biogenesis, innate immune signaling, and R-loop resolution.
PATHOGENESIS AND MUTATIONAL LANDSCAPE OF DDX41-MUTATED MYELOID NEOPLASMS
The pathogenesis of DDX41-related myeloid neoplasms follows the classical tumor suppressor model, wherein inherited loss-of-function germline variants predispose individuals to malignancy by allowing subsequent acquisition of somatic alterations in the remaining functional allele. Clinical cohort studies consistently demonstrate that a substantial proportion of patients with germline DDX41 variants who develop myeloid neoplasms harbor somatic second-hit mutations. The p.R525H missense variant within the helicase domain represents the most frequent somatic second hit, occurring at the critical ATP-binding site and disrupting essential helicase activity [19].
Multiple mechanistic studies have elucidated how DDX41 mutations impair hematopoiesis, providing insights into their contribution to age-related hematopoietic dysfunction and myelodysplasia. Reported mechanisms include defective snoRNA processing and impaired ribosome assembly leading to reduced protein synthesis [30]; erythropoietic defects associated with cell cycle arrest and activation of DNA damage responses [39]; and R-loop–mediated DNA damage driven by dysregulated N6-methyladenosine methylation complexes [27]. Pathogenesis may be further accelerated by synergistic interactions with TP53 mutations, which partially compensate for DDX41 haploinsufficiency while promoting highly penetrant hematologic malignancies resembling MDS and AML [40]. These findings emphasize the intricate interplay between DDX41 dysfunction and hematopoietic regulation, underscoring the need for continued investigation to clarify the molecular mechanisms underlying DDX41-mutated myeloid neoplasms.
The defining features of germline DDX41 variants include their distinctive clustering patterns and their frequent coexistence with somatic DDX41 mutations. Most germline variants manifest as loss-of-function alterations in the N-terminal region, whereas recurrent somatic missense variants are predominantly localized within the C-terminal region. Commonly reported germline alterations include p.M1? (loss of start codon), p.D140fs, and p.A500fs (frameshift variants). In a cohort of 346 patients of diverse ethnic origins carrying germline DDX41 variants, the most prevalent alteration was p.A500fs (23.1%), followed by p.D140fs (11.3%), p.M1? (5.5%), p.S363del (4.3%), and p.Y259C (3.8%) [41]. Nontruncating germline variants were largely concentrated in the DEAD-box domain, whereas nontruncating somatic variants occurred primarily in the helicase domain. Recent analyses from four large cohorts (each including more than 100 patients) investigating germline DDX41 variants are summarized in Table 1 [18, 41–43].
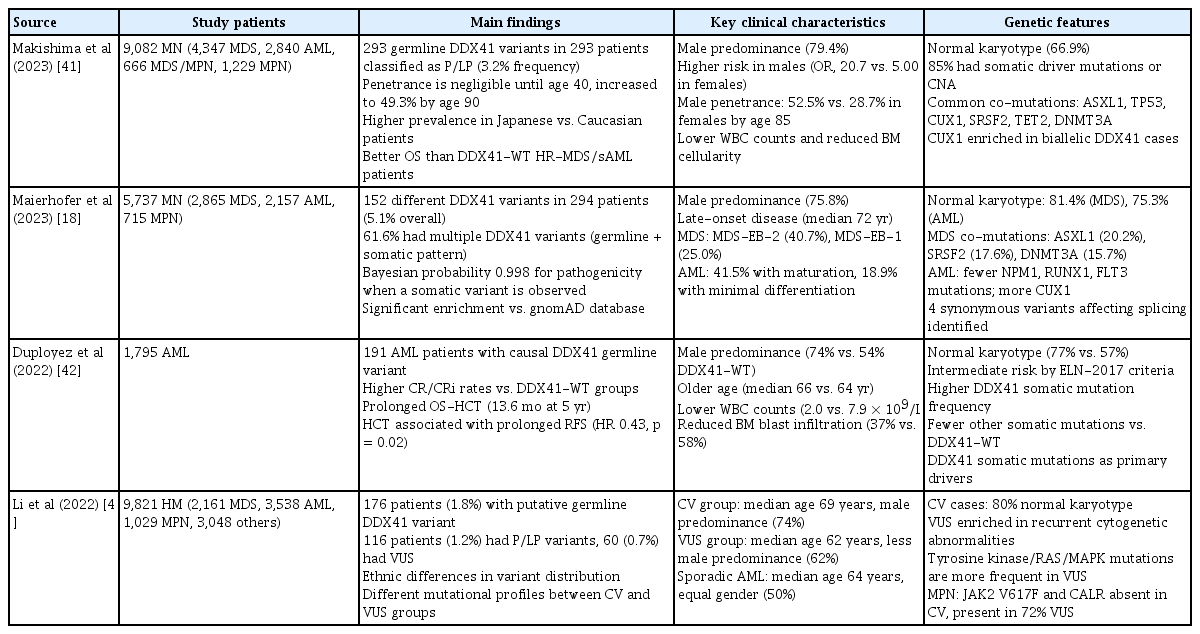
Summary of large cohort studies involving patients with myeloid neoplasms carrying germline DDX41 variants
Significant ethnic variation in the distribution of germline DDX41 variants has been demonstrated in multiple studies, reflecting founder effects and population-specific genetic backgrounds [2,4]. In Asian populations, the variants p.A500fs and p.Y259C, together with the splice-site alteration c.935+4A>T, account for more than 50% of pathogenic germline DDX41 variants. Cohorts from Europe and North America exhibit distinct mutational spectra, with p.M1? and several frameshift variants—particularly p.D140fs—being more prevalent. Notably, p.A500fs, which is most common in Japan, is absent in Western populations, whereas p.D140fs, the predominant germline variant in the United Kingdom, United States, and France, is not observed in Asian populations. Focusing on East Asian populations, four reports have described germline DDX41 variants associated with myeloid neoplasms from Japan, China, and Korea. The predominant variants differ among these countries: p.A500fs is common in Japan, p.Y259C in Korea, and the splicing variant c.935+4A>T in China [27, 41,43,44]. Interestingly, while p.V152G and c.935+4A>T are frequent in Korea and China, they have not been identified in the Japanese population. The E7* variant occurs in a subset of patients in Korea and Japan but has not been reported in China (Fig. 2). These population-specific variant distributions have important implications for genetic testing strategies. Targeted screening panels may require population-tailored optimization to enhance diagnostic sensitivity. Moreover, the founder effects observed in certain populations suggest that cascade testing within families and communities could be particularly valuable for identifying at-risk carriers in regions with high-frequency founder variants.
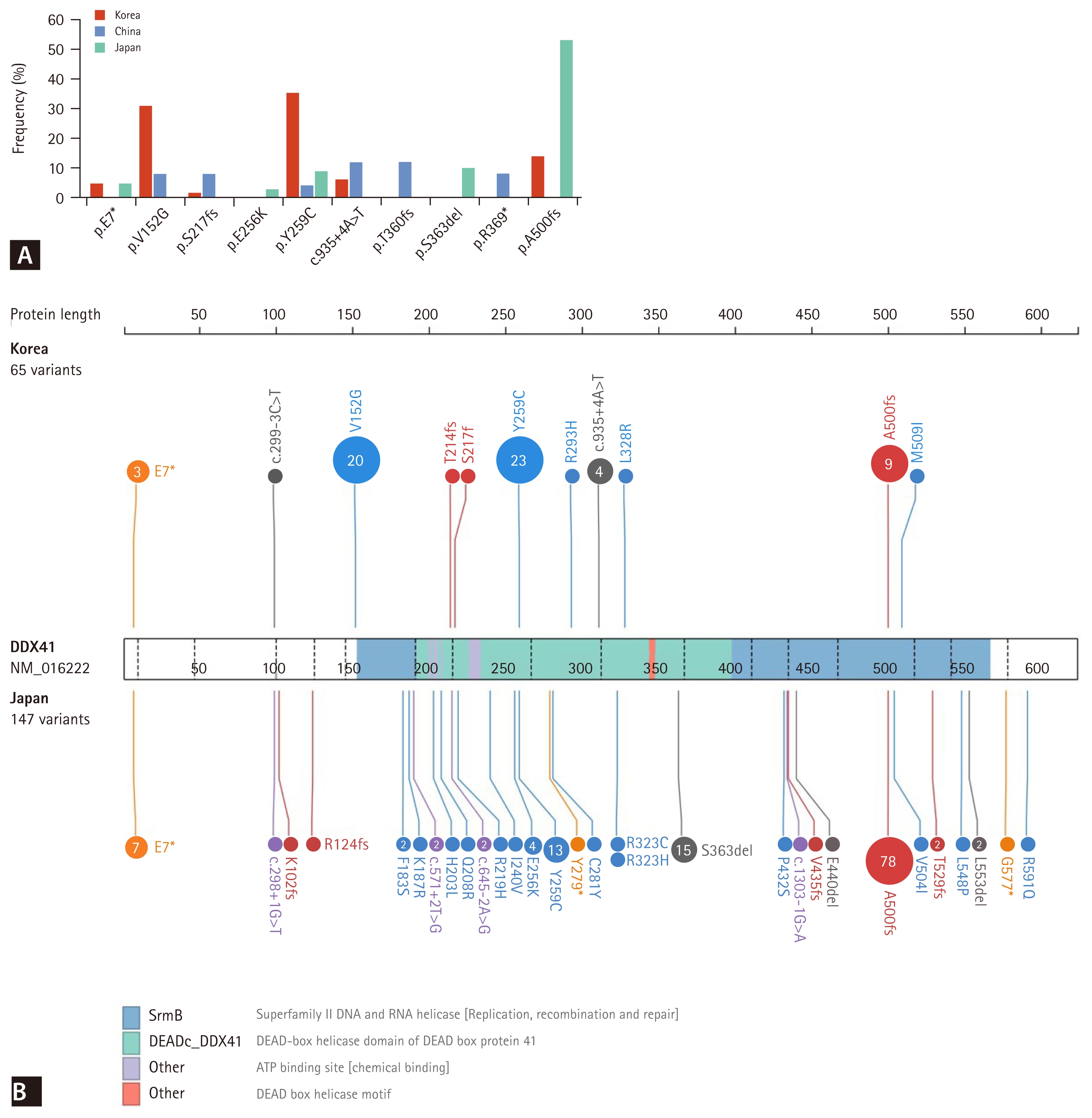
Distribution of germline DDX41 variants in East Asian populations. (A) Frequency plot of recurrent germline DDX41 variants identified in Korean, Chinese, and Japanese cohorts. (B) Distribution of germline DDX41 variants in Korean (upper panel, n = 65) and Japanese (lower panel, n = 147) populations.
It is noteworthy that recent evidence indicates DDX41 variants are relatively common in the general population and are strongly associated with an increased risk of MDS and AML [45]. Analysis of 454,792 participants in the UK Biobank identified 452 distinct nonsynonymous DDX41 variants among 3,538 individuals, corresponding to an overall carrier frequency of approximately 1 in 129 individuals for any nonsynonymous variant. Within the subgroup of carriers harboring pathogenic variants (1,059 individuals), 34 developed myeloid malignancies, yielding an odds ratio of 12.3 for MDS/AML compared with noncarriers. Consistent with observations from clinical cohorts, European populations demonstrated a higher prevalence of specific variants—such as p.M1? and p.D140fs—that were absent in individuals of non-European ancestry. Moreover, population-based analyses indicate that DDX41 carriers do not exhibit an increased frequency of clonal hematopoiesis relative to noncarriers, suggesting that DDX41-related leukemogenesis may proceed through distinct evolutionary pathways that culminate directly in overt malignancy rather than gradual clonal expansion. Collectively, these findings identify DDX41 as one of the most common cancer predisposition genes in the general population, underscoring its relevance to population screening and genetic counseling strategies.
CLINICAL MANIFESTATIONS OF DDX41-ASSOCIATED MYELOID NEOPLASMS
DDX41-associated myeloid neoplasms exhibit a characteristic late-onset presentation with marked male predominance, distinguishing them from other hereditary predisposition syndromes. Clinical cohorts consistently report median ages at diagnosis clustering between the late sixth and early seventh decades of life. Two Korean cohorts comprising 39 and 28 patients showed median ages of 63 and 66 years, respectively [27, 44]; an international cohort of 431 patients reported a median age of 69 years [46]; and multiple other referral-based studies confirmed median ages of 69–70 years [47]. The penetrance of myeloid neoplasms associated with germline DDX41 variants has not yet been fully determined [10]. Among carriers of germline DDX41 variants, the penetrance of any myeloid neoplasm is minimal before age 40 but rises sharply to 49–54% by age 90 [41,48]. The development of MDS and AML occurs more frequently in males than in females, with an approximate 3:1 ratio [19, 47,49].
Germline DDX41 variants are associated with a broad spectrum of myeloid neoplasms, with MDS and AML being the most common manifestations. In a Korean study of 457 patients with AML, MDS, and idiopathic cytopenia of undetermined significance (ICUS), germline DDX41 variants were identified in 4 (2.3%) of 172 patients with AML, 19 (9.0%) of 210 with MDS, and 5 (6.7%) of 75 with ICUS [44]. Similarly, a large international cohort of 9,082 patients revealed DDX41 variants in 346 patients (3.8%), including 201 (4.6%) of 4,347 patients with MDS, 136 (4.8%) of 2,840 with AML, 1 (0.08%) of 1,229 with myeloproliferative neoplasms (MPN), and 8 (1.2%) of 666 with MDS/MPN overlap syndromes [41].
The acquisition of somatic second hits—most commonly the p.R525H variant—represents a key molecular event influencing disease subtype and progression [19, 42]. Studies have shown that somatic DDX41 variants occur in approximately 60–80% of AML or MDS cases with germline DDX41 variants, and more than 95% of these somatic variants are missense [20, 50]. Somatic mutations in genes other than DDX41 occur at lower frequencies compared with patients lacking germline DDX41 variants, with commonly mutated genes including ASXL1, TP53, CUX1, SRSF2, TET2, and DNMT3A [17,20, 41,42,47]. Variant interpretation generally follows the guidelines of the American College of Medical Genetics and Genomics (ACMG) [51], which can result in overclassification of DDX41 variants as variants of uncertain significance (VUS). However, recent studies have adopted a modified classification approach in which the presence of somatic DDX41 variants serves as supportive evidence for germline pathogenicity [17,18, 52].
DDX41-associated myeloid neoplasms often present with hypocellular bone marrow and lower leukocyte counts at diagnosis, in contrast to the hypercellular marrow and elevated leukocyte counts typically observed in sporadic MDS and AML [17, 41,44, 52]. Furthermore, many patients harboring DDX41 variants experience a subacute clinical course marked by persistent cytopenia that fulfills the criteria for ICUS or clonal cytopenia of undetermined significance (CCUS) before overt myeloid neoplasm development [17,20, 44, 52–54]. Cytogenetic analyses frequently reveal normal karyotypes in DDX41-associated disease, with 60–80% of patients exhibiting normal cytogenetics—a substantially higher rate than in sporadic myeloid neoplasms [20,27, 41,44, 52].
TREATMENT AND PROGNOSIS
MDS and AML harboring DDX41 variants are generally managed with the same disease-directed approaches used for sporadic cases, with multiple retrospective studies reporting high response rates to standard regimens and notable benefit from lower-intensity venetoclax-based combinations in older or unfit patients (Table 2). Treatment selection is guided by disease subtype (MDS versus AML), patient fitness, donor availability, and the presence of co-mutations or adverse cytogenetic features. The high prevalence of normal cytogenetics, together with the distinctive molecular signature of DDX41 variants, has led to recognition that conventional cytogenetic risk stratification models may not accurately predict outcomes in these patients [20, 42]. Current prognostic scoring systems—specifically, the International Prognostic Scoring System–Molecular for MDS [55] and the European LeukemiaNet (ELN) 2022 classification for AML [10] —do not include DDX41 status. However, the recently introduced ELN 2024 Less-Intensive model for AML, which incorporates venetoclax-based regimens, categorizes DDX41-mutated AML as favorable risk [56,57]. Despite these favorable outcomes with non-transplant regimens, patients with DDX41-mutated MDS or AML remain at risk of relapse or disease progression without allogeneic hematopoietic cell transplantation (HCT) [42, 58].
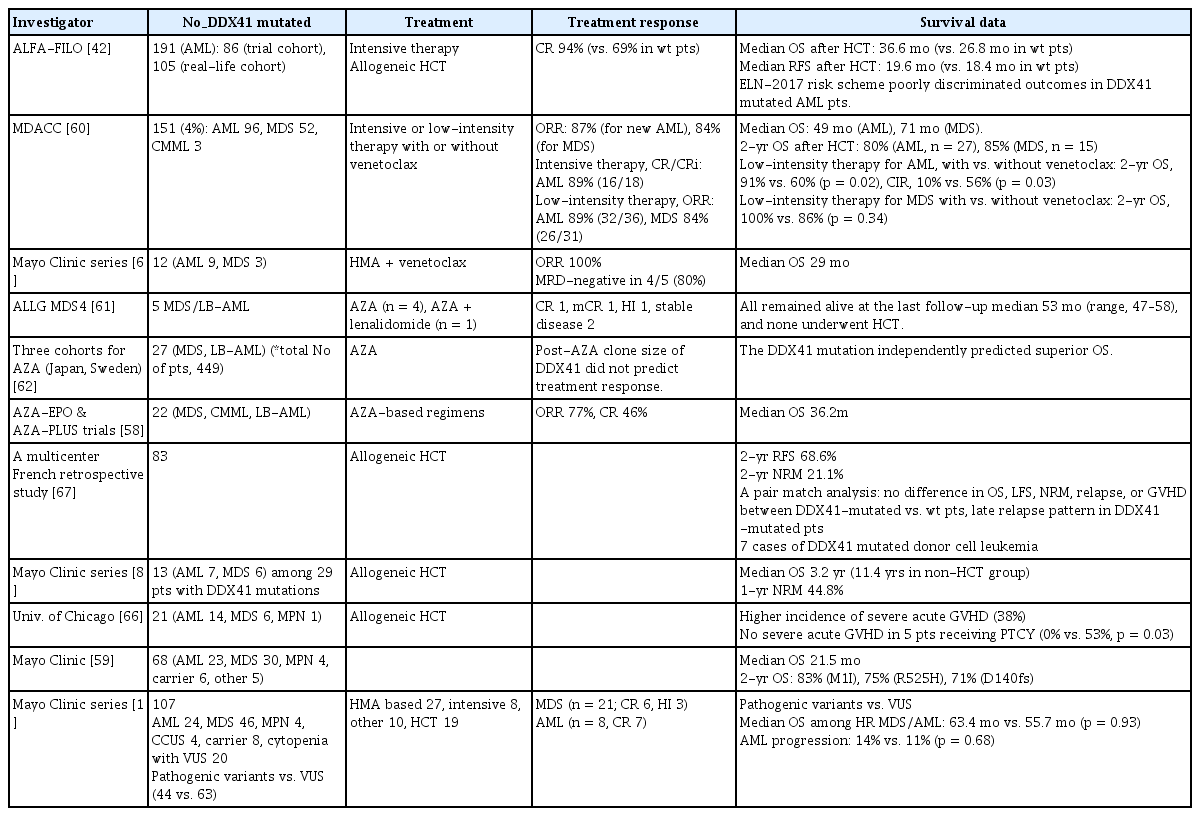
Treatment responses and survival outcomes in patients with DDX41-mutated myeloid neoplasms
High response rates to intensive chemotherapy have been observed in DDX41-mutated patients. In a large single-center cohort, complete remission (CR) rates following intensive chemotherapy were 89% in newly diagnosed and 67% in relapsed or refractory DDX41-mutated AML patients [59]. Another study compared outcomes of 191 newly diagnosed patients with DDX41-mutated AML to those of similarly treated patients without DDX41 variants [42]. All patients received intensive chemotherapy; those with DDX41-mutated AML achieved higher CR rates (94% vs. 69%, p < 0.0001) and longer overall survival (p < 0.001) than ELN intermediate/adverse-risk patients lacking DDX41 variants. The ELN 2017 risk classification failed to effectively discriminate long-term outcomes among DDX41-mutated AML patients in this analysis [42].
Favorable outcomes have also been reported following low-intensity therapy with hypomethylating agents in patients with DDX41-mutated AML or MDS. The overall response rates after low-intensity therapy, with or without venetoclax, were 89%, 46%, and 84% for previously untreated AML, relapsed/refractory AML, and MDS with DDX41 variants, respectively [60]. In a small cohort, three of five patients treated with azacitidine, with or without lenalidomide, achieved responses—CR in one, marrow CR in one, and hematologic improvement in one. All five patients remained alive at the last follow-up (median, 53 mo; range, 47–58 mo) without allogeneic HCT [61]. A post hoc analysis of two clinical trials demonstrated favorable and durable responses to azacitidine-based regimens in 22 patients with DDX41-mutated higher-risk MDS and low-blast AML, with an overall response rate of 77%, CR of 46%, and median overall survival of 36.2 months [58]. In a separate study of three independent cohorts of patients with MDS and related myeloid neoplasms treated with azacitidine, the presence of DDX41 variants independently predicted superior overall survival [62]. DDX41-mutated AML and MDS are known to exhibit a relatively low mutational burden compared with unselected cohorts [41, 52], and this reduced mutational complexity may contribute to improved responses to hypomethylating therapy [63]. Among AML and MDS patients treated with low-intensity regimens, the addition of venetoclax was associated with improved survival and lower relapse rates [60]. In a small series of 12 patients with DDX41-mutated AML (n = 9) or MDS (n = 3) treated with a hypomethylating agent plus venetoclax, all patients responded, with four of five evaluable cases achieving minimal residual disease negativity [64]. Several molecular and clinical studies have reported higher-than-expected responses to lenalidomide in patients harboring DDX41 variants, although these findings are limited by small sample sizes and retrospective design [19, 65].
In a single-institution retrospective analysis, DDX41 variants were associated with a 38% incidence of severe (grade III–IV) acute graft-versus-host disease (GVHD) after allogeneic HCT among 21 patients with myeloid neoplasms. Notably, none of the five patients receiving post-transplant cyclophosphamide developed severe acute GVHD [66]. A multicenter French retrospective study including 83 AML and MDS patients with DDX41 variants found no higher incidence of severe acute GVHD and no significant differences in post-transplant outcomes compared with patients without DDX41 variants [67]. This study also reported seven cases of donor cell leukemia with DDX41 mutations, consistent with earlier case reports [68,69]. Approximately 0.1–0.5% of post-transplant malignancies originate from transplanted donor cells, and such donor-derived leukemias may arise from germline predisposition alleles or donor clonal hematopoiesis [70]. For patients undergoing allogeneic HCT who harbor deleterious germline DDX41 variants, related donors should undergo targeted genetic testing before donation [70,71].
The presence of DDX41 variants is associated with a generally favorable prognosis in multiple studies; however, clinical outcomes are influenced by disease subtype, variant type, and classification. A recent meta-analysis pooling available data showed that DDX41 variants correlate with improved overall survival compared with DDX41 wild type in AML (pooled hazard ratio, 0.60; p = 0.008) but not in MDS (pooled hazard ratio, 0.97; p = 0.95) [72]. In a single-center cohort of 68 patients with DDX41 variants, three major hotspots—p.M1, p.R525H, and p.D140fs—were associated with 2-year overall survival rates of 83%, 75%, and 71%, respectively [59]. Molecular hematology laboratories generally apply ACMG criteria, leading to classification of many DDX41 variants as VUS. In one study, patients with VUS had similar molecular and clinical characteristics to those with pathogenic DDX41 variants [16], underscoring the challenges of interpretation and the need for careful germline assessment [17,18, 52].
DDX41 VARIANTS IN NON-MYELOID NEOPLASMS
Studies involving individuals with germline DDX41 predisposition indicate that non-myeloid neoplasms do occur but remain uncommon. In a cohort of 195 patients with germline DDX41 variants, clinical diagnoses included asymptomatic carriers (n = 20, 10.2%), CCUS (n = 12, 6.1%), clonal hematopoiesis (n = 10, 5.1%), MPN (n = 13, 6.7%), MDS (n = 79, 40.5%), AML (n = 40, 20.5%), chronic myelomonocytic leukemia (n = 2, 1.0%), mast cell leukemia (n = 1, 0.5%), lymphoid neoplasms (9.2%), plasma cell dyscrasias (6.1%), and solid tumors (22.5%) [17]. Some studies of myeloid neoplasms with DDX41 variants have also reported a subset of patients with personal or family histories of lymphoid or solid malignancies [73,74]. In a comprehensive investigation involving 93 individuals from 43 families carrying germline DDX41 variants, both solid cancers and hematologic malignancies were observed. The estimated penetrance among individuals with germline DDX41 variants was 54% (n = 50) for hematologic malignancies by age 90 years and 24% (n = 22) for solid tumors by age 75 years [48].
GENETIC COUNSELING AND CLINICAL APPLICATION
Healthy carriers of pathogenic germline DDX41 variants pose unique challenges for genetic counseling due to incomplete penetrance, late disease onset, and variant heterogeneity. This highlights the importance of structured surveillance with age-stratified monitoring, functional assessment of uncertain variants, and cascade testing of at-risk relatives [75–77]. Current evidence indicates that while NGS–based panels may incidentally detect such variants, comprehensive germline testing and individualized counseling remain essential for optimizing risk communication, clinical management, and long-term outcomes in carriers of DDX41-associated predisposition [78,79]. Healthcare professionals should explain that the disease typically manifests after age 50 years, with many patients presenting with preceding cytopenias or leukopenia before overt malignancy develops [16, 59]. Genetic counseling sessions should also address the psychological impact of a positive result, including anxiety about cancer risk, implications for family members, and concerns regarding insurance coverage or employment discrimination.
The clinical management of DDX41 germline variant carriers involves baseline complete blood count evaluation upon confirmation of carrier status, followed by individualized periodic hematologic monitoring with a low threshold for hematology referral when new cytopenias or abnormal blood count trends emerge [9, 80]. Family cascade testing is crucial given the autosomal dominant inheritance pattern, as first-degree relatives have a 50% probability of carrying the variant and should be offered predictive genetic testing [16, 80].
A critical component of DDX41 genetic counseling involves addressing the implications for allogeneic HCT, both for affected individuals requiring treatment and for potential related donors. For patients with DDX41-associated myeloid neoplasms who are candidates for allogeneic HCT, rapid predictive testing of potential related donors is essential to exclude carriers, as use of a DDX41-positive donor carries a measurable risk of donor-derived malignancy [68–70]. Counseling should emphasize early donor screening, consideration of alternative donor sources when family members are carriers, and ongoing communication regarding genetic test results and surveillance recommendations within the family.
FUTURE DIRECTIONS
The field of DDX41 variant research is advancing rapidly through large-scale genomic sequencing initiatives and sophisticated functional studies that continue to clarify the pathogenic mechanisms driving myeloid neoplasms. These discoveries are being incorporated into improved variant classification frameworks that enhance diagnostic precision and clinical interpretation. Mechanistic insights into the role of DDX41 in both its functional and dysfunctional states are identifying novel therapeutic targets and opportunities for precision medicine, although most strategies remain in the preclinical phase. Direct modulation of RNA helicases represents a particularly promising therapeutic avenue, as DEAD-box helicases possess druggable surfaces and exhibit synthetic lethal interactions with other helicases that may be exploited for therapeutic benefit [81]. The marked splicing abnormalities resulting from DDX41 dysfunction suggest that splicing-directed therapies—such as small molecules that modulate spliceosome activity or correct specific splicing defects—may offer targeted treatment strategies for DDX41-associated malignancies [35, 82]. Furthermore, the accumulation of R-loops and subsequent genomic instability in DDX41-deficient cells indicate that agents enhancing R-loop resolution or preventing their formation could represent viable therapeutic options [26]. Modulation of the DNA damage response constitutes another potential therapeutic strategy, as DDX41-deficient cells display altered dependencies in DNA repair pathways that can be exploited through synthetic lethality. Poly (ADP-ribose) polymerase inhibitors and other DNA damage response modulators may demonstrate increased efficacy in DDX41-associated malignancies due to persistent replication stress and defective DNA repair [83]. The development of therapeutic strategies specifically targeting DDX41 will require close collaboration among basic researchers, translational scientists, and clinicians to effectively translate promising preclinical discoveries into clinical applications.
The late-onset and frequently indolent nature of DDX41- associated myeloid neoplasms presents distinct opportunities for preventive measures and early intervention strategies aimed at delaying or preventing malignant transformation in at-risk individuals [13, 45, 53]. Integrating genetic risk assessments, family history data, and molecular biomarkers could enable personalized surveillance programs that balance the benefits of early detection against the financial, psychological, and clinical burdens of intensive monitoring. Prospective registries and longitudinal cohort studies remain essential to validate these preventive and early intervention approaches, as they will provide natural history data critical for designing and powering interventional trials. The relatively high prevalence of DDX41 variants in the general population, combined with their typically late-onset phenotype, offers a compelling rationale for population-based screening initiatives and preventive trials with potential public health benefits. Future research priorities should include the development of standardized biomarker panels, validation of early intervention approaches in prospective cohorts, and the establishment of international collaborative networks to accelerate scientific progress and facilitate rapid clinical translation of emerging discoveries.
CONCLUSION
The identification of DDX41 as a key hereditary cancer predisposition gene has profoundly transformed the understanding of adult-onset myeloid malignancies and redefined the framework for hereditary hematopoietic disorders. The distinct clinical and molecular characteristics of DDX41-associated neoplasms—including late-onset presentation, relatively favorable prognosis, and unique pathogenic mechanisms—demand tailored management strategies integrating genetic counseling, structured surveillance, and personalized treatment approaches. As comprehensive genomic sequencing efforts continue to expand insight into DDX41 variant pathogenicity and functional impact, the field is increasingly positioned to design targeted therapeutic and preventive interventions for at-risk individuals and their families. Future investigations should aim to elucidate the molecular pathways of DDX41-mediated malignant transformation, optimize surveillance protocols, and develop precision therapies to improve patient outcomes.
Notes
Acknowledgments
We got some assistance from an AI program (SciSpace) in English editing of our manuscript and drafting in the section of “FUTURE DIRECTIONS.”
CRedit authorship contributions
Je-Hwan Lee: conceptualization, methodology, resources, investigation, writing - original draft, writing - review & editing, supervision; Eun-Hye Hur: conceptualization, methodology, resources, investigation, writing - original draft, writing - review & editing; Young-Uk Cho: conceptualization, methodology, resources, investigation, writing - original draft, writing - review & editing
Conflicts of interest
The authors disclose no conflicts.
Funding
None