Long-term outcome of interstitial lung disease in patients with primary Sjögren’s syndrome: a retrospective observational study
Article information

Abstract
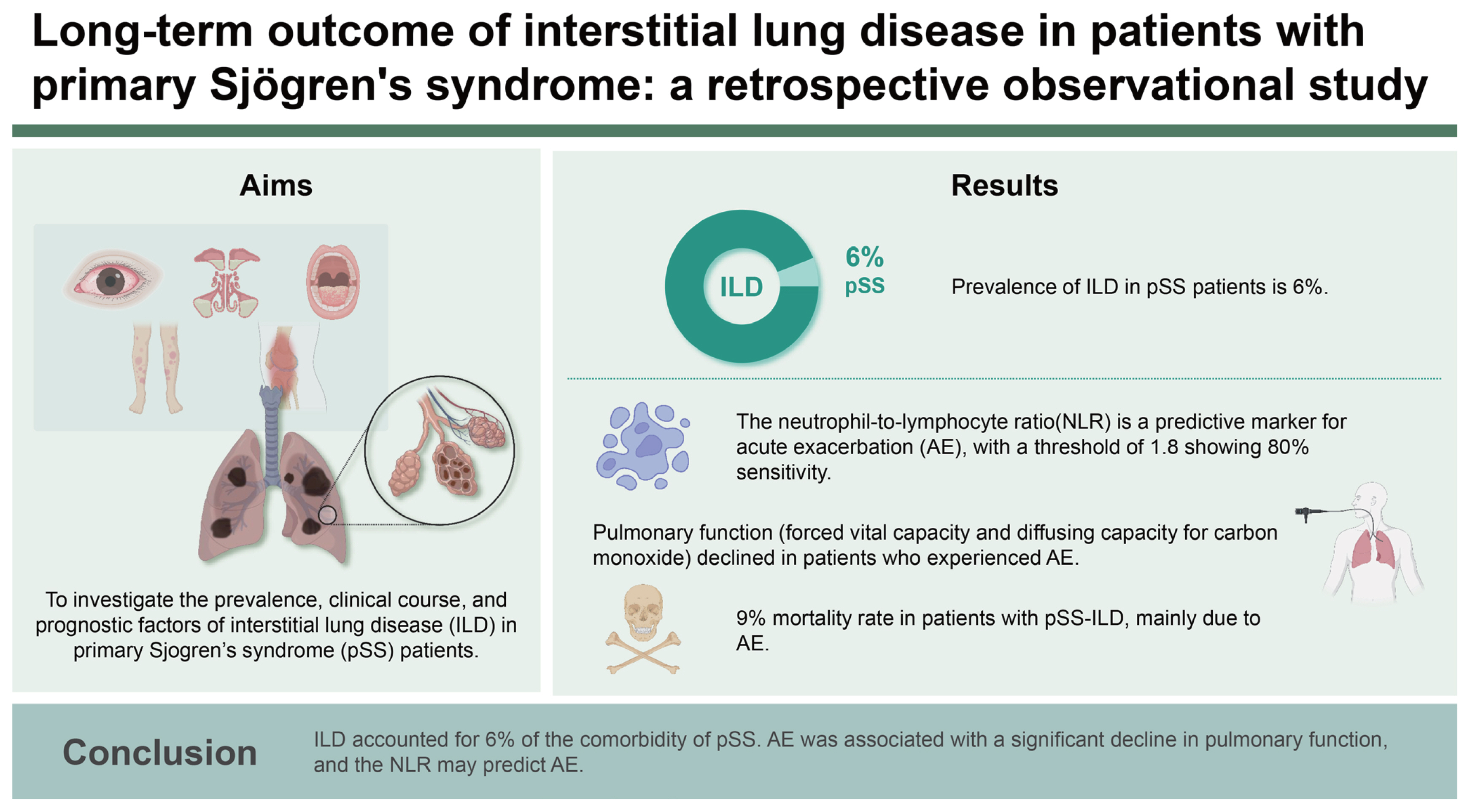
Background/Aims
Interstitial lung disease (ILD) is a potentially serious but underdiagnosed manifestation of primary Sjögren’s syndrome (pSS). This observational study investigated the prevalence and clinical course of ILD in pSS, together with prognostic factors.
Methods
A multicenter, retrospective longitudinal study was performed using findings from baseline and follow-up pulmonary function tests and chest computed tomography. Predisposing factors for the development of ILD and acute exacerbation (AE) were identified using a logistic regression model. The risk factors for a significant decline of pulmonary function were determined by the Cox proportional hazard model.
Results
A total of 1,306 patients with pSS were included in this study (female, 98%; mean age, 54 years). Among them, 79 patients with pSS were comorbid with ILD. ILD was more frequently found in male, older patients. Nonspecific interstitial pneumonia was the most prevalent imaging pattern in pSS-ILD (51%), followed by usual interstitial pneumonia (22%). At diagnosis with pSS-ILD, 54% of patients had restrictive pulmonary function, and 41% of patients initiated pharmacological treatment. During the median 4-year follow-up period, AE, a significant decline in pulmonary function, and death occurred in 19%, 29%, and 9% of patients with pSS-ILD, respectively. The neutrophil-to-lymphocyte ratio (NLR) increased 3 months prior to AE, and it was associated with AE. Older age at pSS-ILD diagnosis was a prognostic factor for a significant decline in pulmonary function.
Conclusions
ILD accounted for 6% of the comorbidity of pSS. AE was associated with a significant decline in pulmonary function, and the NLR may predict AE.
INTRODUCTION
Primary Sjögren’s syndrome (pSS) is a systemic autoimmune disease characterized by lymphocytic infiltration of the salivary and lacrimal glands. The incidence of pSS is estimated at 4–7 per 100,000 people, and the prevalence is estimated at 2–3% [1,2]. The incidence is relatively high among those with autoimmune connective tissue diseases, followed by those with rheumatoid arthritis [3].
Interstitial lung disease (ILD) is a potentially serious but underdiagnosed manifestation of pSS. The pulmonary involvement of SS includes airway abnormalities, interstitial pneumonia, and lymphoproliferative disorders [4]. The prevalence of ILD in pSS (pSS-ILD) is heterogeneous, ranging between 10% and 20%, depending on ethnicity, the definition of ILD, and pSS [5,6].
In general, people with pSS can expect to live a normal lifespan. However, ILD in pSS is associated with premature mortality [7]. The 10-year survival rate for pSS-ILD has been reported to be 79–82%, and the most predominant cause of death was respiratory failure after acute exacerbation (AE) or chronic progression of ILD [8–10]. Identifying the predisposing factors for AE and a decline in pulmonary function is crucial to reducing mortality and comorbidities.
To date, only a limited number of studies have evaluated the long-term progression of pSS-ILD as well as its associated prognostic factors. We analyzed the prevalence of ILD in patients with pSS admitted to three university-affiliated hospitals in Korea over a 10-year period and identified the underlying factors that contribute to the development of ILD in pSS patients. In addition, we investigated the natural course of pSS-ILD by tracing the changes in pulmonary function tests (PFTs) and serial computed tomography (CT) scans of the chest. As part of our investigation, we also identified predisposing factors for AE and a significant decline in lung function.
METHODS
Patients and study design
This was a multicenter retrospective longitudinal study of patients with SS treated at Seoul St. Mary’s Hospital, Bucheon St. Mary’s Hospital, or Uijeongbu St. Mary’s Hospital in Korea. The data were extracted from the clinical data warehouse (CDW) of the Catholic Medical Center. Using the International Classification of Diseases (ICD) 10th revision, patients with pSS from March 1, 2012, to February 28, 2022, were assigned code M35.0. In Korea, patients with pSS who fulfill the 2002 American-European Consensus Group classification criteria [11] and/or 2016 American College of Rheumatology/European League Against Rheumatism classification criteria [12] are registered in the rare and incurable diseases medical care assistance program of the Korean Ministry of Health and Welfare. To register this assistant program, rheumatologists should confirm whether the classification criteria are met. To reduce the potential errors that may arise when selecting patients through ICD code, we also applied this registration code for identifying patients with pSS (Fig. 1A). Patients were excluded if they had a second autoimmune disease code (M05.8 and M05.9 for seropositive rheumatoid arthritis; M32 for systemic lupus erythematosus; M33.0 and M33.1 for dermatomyositis; M33.2 for polymyositis; M33.9 for dermatopolymyositis; M34.0, M34.1, M34.8, and M34.9 for systemic sclerosis; M35.1 for mixed connective tissue disease; M35.2 for Behcet’s disease; M35.2 for polymyalgia rheumatica; M35.6 for relapsing panniculitis). In addition, patients who were positive for anti-centromere, anti-double strand DNA, anti-Jo1, or anti-RNP antibodies were excluded, due to potential overlap syndrome. The CDW provides anonymized access to electronic health records, imaging studies, and PFTs. The identification of patients with pSS was ascertained through a review of electronic health records.
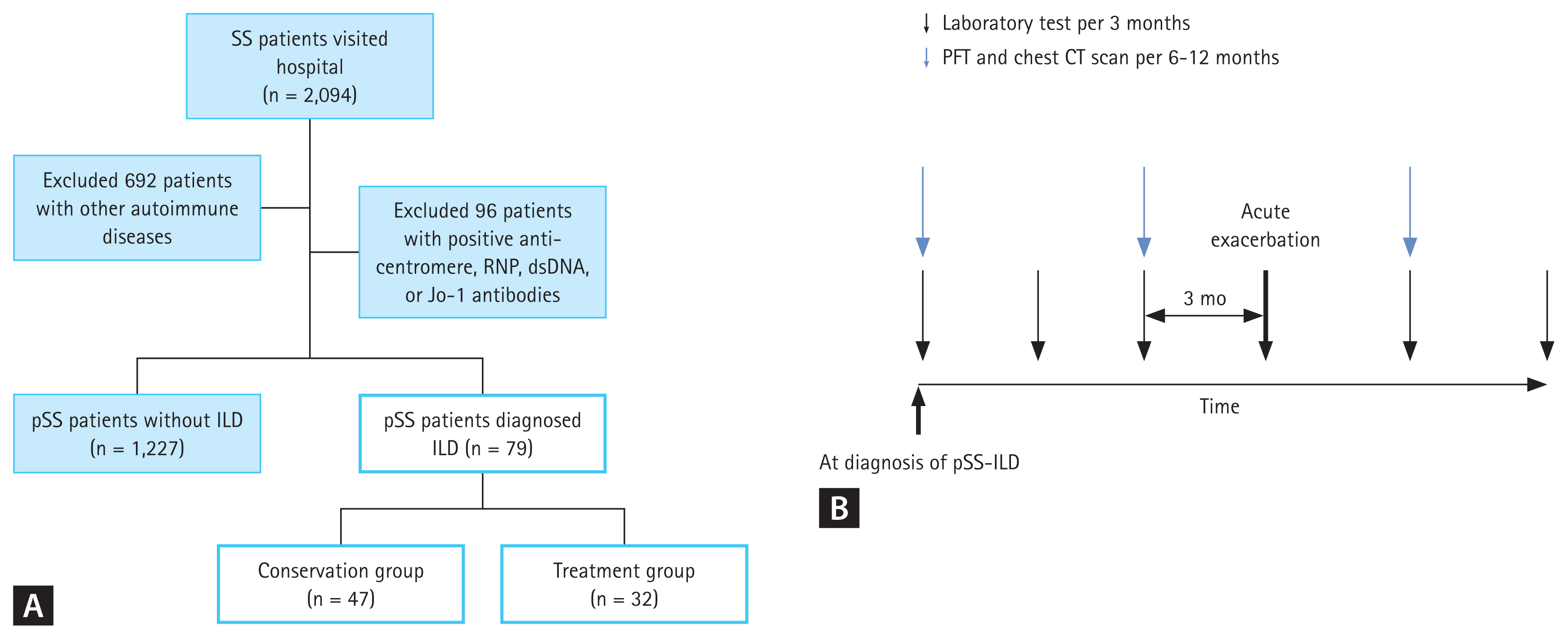
Flowchart of patients screening and classification (A) Application of selection criteria and patient disposition. (B) An overview of clinical data collection over time. Patients with pSS-ILD used to assess laboratory examination per three months, PFT, and chest CT scan per 6 to 12 months. The bold arrow indicates the time of acute exacerbation, and the laboratory test results from 3 months and 6 months before acute exacerbation were reviewed. SS, Sjögren’s syndrome; pSS, primary Sjögren’s syndrome; ILD, interstitial lung disease; PFT, pulmonary function test; CT, computed tomography.
The study protocol was approved by the Institutional Review Board of the Catholic Central Medical Center (UC22WIDE0042). Informed consent was waived, as the study was retrospective.
Evaluation of ILD
All patients with pSS who underwent chest CT scans were reviewed. Pulmonary high-resolution CT (HRCT) images were obtained at initial ILD diagnosis in 53 patients (67%) with 1.0 or 1.5 mm–thick sections throughout the entire lungs. The other 26 patients (33%) underwent conventional chest CT with 1.0 or 2.5 mm-thick sections. The chest CT findings were evaluated by board-certified thoracic radiologists in each hospital. We defined ILD based on chest CT reports of ILD, interstitial pneumonia with autoimmune features, idiopathic interstitial pneumonia, usual interstitial pneumonia (UIP), non-specific interstitial pneumonia (NSIP), respiratory bronchiolitis-ILD, organizing pneumonia (OP), acute interstitial pneumonia, desquamative interstitial pneumonia, and lymphoid interstitial pneumonia [13], as well a recorded diagnosis. In addition, patients who underwent lung biopsy with results consistent with ILD were also classified as having ILD [7]. No cases had an identifiable cause for ILD other than underlying pSS.
Patients diagnosed with pSS-ILD were categorized into two groups: the treatment group and the conservative group. In the treatment group, patients who commenced treatment with glucocorticoids (at a prednisolone equivalent dose ≥ 30 mg/day) and/or immunosuppressive agents (cyclophosphamide, mycophenolate mofetil [MMF], and azathioprine) following the ILD diagnosis. The conservative group encompassed patients who did not undergo specific ILD treatment. This group comprised individuals who were already on glucocorticoids or immunosuppressants for managing other symptoms before the ILD diagnosis, confirmed through chart reviews. Those whose glucocorticoids dosage was escalated to 30 mg/day or higher after the ILD diagnosis were categorized as the treatment group.
Data collection
We collected demographic data (date of birth and sex), date of pSS diagnosis, date of ILD diagnosis, date of last follow-up, status at the last follow-up (dead or alive), and comorbidity details. Laboratory features recorded at diagnosis were the serological profiles of antinuclear antibody, anti-Ro/Sjögren’s syndrome-A (SSA), anti-La/Sjögren’s syndrome-B (SSB), rheumatoid factor, and complete blood counts (CBCs). The neutrophil-to-lymphocyte ratio (NLR) is measured from a CBC. Between March 2012 and June 2019, anti-Ro/SSA and anti-La/SSB antibodies were detected in patients at Seoul St. Mary’s Hospital using an enzyme immunoassay (EIA), since June 18, 2019, a line immunoassay (ImmunoBlot) has been employed. Anti-Ro/SSA and anti-La/SSB antibodies were also detected using EIA at Bucheon and Uijeongbu St. Mary’s Hospital throughout the study period.
Pulmonary function indices (forced vital capacity [FVC] and diffusing capacity for carbon monoxide [DLCO]) at the time of diagnosis of ILD and subsequent follow-up were reviewed.
Clinical outcomes of pSS-ILD
An AE was defined as a clinically significant respiratory worsening requiring hospitalization, particularly new or worsening ground-grass opacity and/or consolidation on CT scans [14]. Cases with documented respiratory pathogens during hospitalization were excluded from the AE classification. In addition, cases primarily treated with antibiotics and discontinued or reduced immunosuppressants and/or glucocorticoids were also excluded. To determine whether there were predictive factors for AE, laboratory and therapeutic records from 3 and 6 months before an AE event were collected (Fig. 1B). Survival status and cause of death were also recorded.
The long-term clinical outcomes of pSS-ILD were grouped into three categories: patients who experienced an AE during follow-up (AE), those who maintained a stable condition without treatment (stable), and those who received treatment from the onset of the disease and did not experience an AE (improvement).
The course of ILD was evaluated by changes in pulmonary function indices. Considering the variations in follow-up intervals among patients, annual changes in FVC and DLCO were calculated by normalizing the difference at each follow-up interval (For example, FVCfollow-up - FVCbaseline/follow-up interval). In the AE group, annual changes were determined from the first occurrence of AE. Meanwhile, for the improvement and stable groups, annual changes were computed from baseline FVC and DLCO. A cumulative decline in FVC greater than 10% or in DLCO greater than 15% from baseline values was considered a significant change in lung function [15]. A marginal decline in lung function was defined as a reduction from baseline value of 5–10% for FVC or 7.5–15% for DLCO [16].
Statistical analysis
Demographic and disease characteristics are expressed as mean ± standard deviation, or as a percentage. PFT results, chest CT findings, and serological profiles were not available for all patients, and all analyses were carried out using observed data, with pairwise deletion of missing values.
T-test was used to compare groups of continuous variables, while the χ2 test and Fisher’s exact test were used to compare categorical variables as appropriate. Changes in FVC and DLCO were calculated as the difference from the baseline using a paired t-test.
The impact of clinical factors associated with ILD development was assessed using binary logistic regression analyses. Model assumptions were tested using residual analysis. To mitigate multicollinearity among variables in the multivariate regression model, the variance inflation factor was assessed. Selection of the most appropriate regression model was based on the minimization of the akaike information criterion. The findings of these analyses are presented as odds ratios (ORs), along with 95% confidence intervals (CIs). Kaplan–Meier plots were used to illustrate the survival curves for pSS with or without ILD, accompanied by a log-rank test for comparison. Survival time was calculated from the date of pSS diagnosis to death or the last follow-up visit (censoring). Moreover, to determine NLR cut-off values for AE prediction, receiver-operator curve analysis was employed, yielding the area under the curve (AUC).
Unadjusted or multivariable Cox proportional hazard models were used to analyze risk factors for a significant decline in lung function. Observational time was calculated from the diagnosis of ILD to the last follow-up visit. Variables with a p value < 0.1 in the unadjusted analysis were entered into multivariable models with backward selection.
All p values were two-sided, with p < 0.05 considered statistically significant. SAS 9.4 software (SAS, Cary, NC, USA) was used for data analysis, and graphs were drawn using Graph-Pad Prism 9.5 (GraphPad Software, San Diego, CA, USA).
RESULTS
Patient characteristics
This study included 1,306 patients with pSS. The mean age at pSS diagnosis was 51 ± 13, and 97.6% were female. The mean follow-up duration after diagnosis of pSS was 6.7 ± 5.2 years. We identified 79 patients with pSS (6%) who were comorbid with ILD (Table 1). Among them, 55 patients (69.6%) had ILD at the time of diagnosis of pSS, and 24 patients (30.4%) developed ILD at a 5.8 ± 3.7 years after diagnosis of pSS.
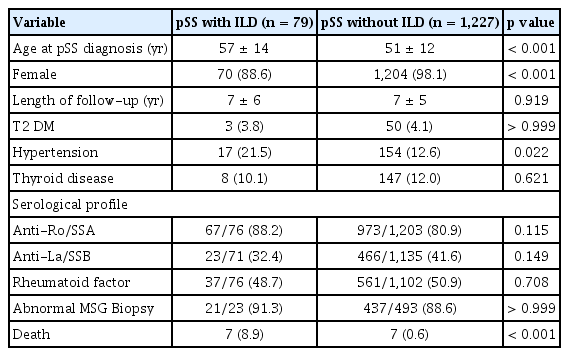
Baseline characteristics of patients with pSS
Characteristics of pSS-ILD
Complete clinical and serologic data were available for most of pSS-ILD subjects and were compared to those of non-ILD patients. Patients with pSS-ILD were diagnosed with pSS at an older age compared with those without ILD. Hypertension was more common comorbidities in patients with pSS-ILD than in the non-ILD patients (Table 1). Multivariate analysis found that age, male sex, and anti-Ro antibody were predictors for ILD (Fig. 2A).
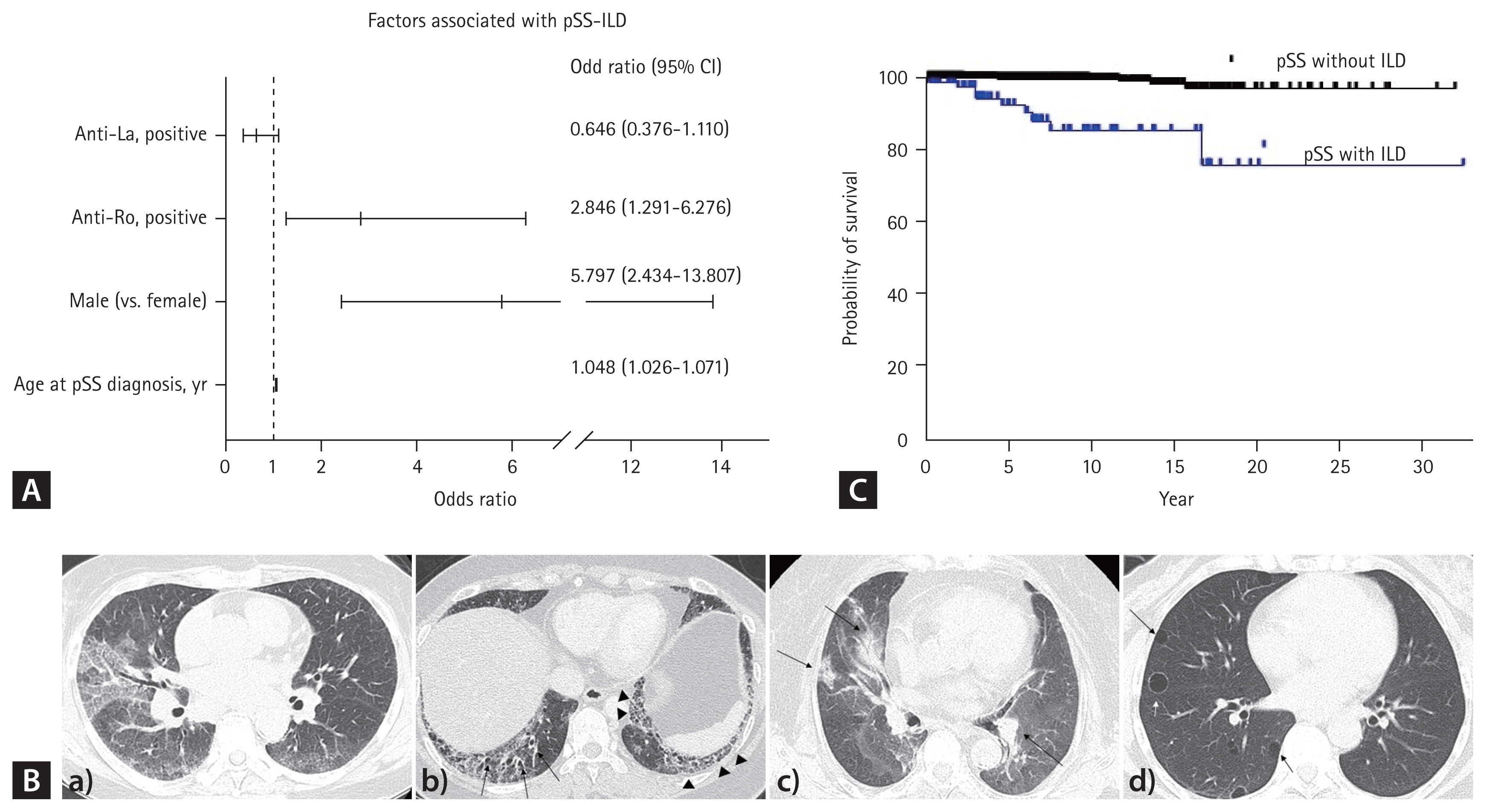
Factors associated with ILD in patients with pSS (A), examples of chest computed tomography images (B), and survival curves for pSS patients with and without ILD (C). a) Non-specific interstitial pneumonia: diffuse peripheral coarse GGOs are evident throughout the entire lung with lower lobar predominance, accompanied by smooth inter/intralobular septal thickening in a patient who experienced acute exacerbation. b) Usual interstitial pneumonia: the image displays honeycomb (arrowhead) with traction bronchiectasis (arrows), combined with subpleural GGOs. c) Cryptogenic organizing pneumonia: characterized by multiple consolidations, some with air bronchograms, distributed bilaterally in both lungs (arrows). d) Lymphocytic interstitial pneumonia: multiple, various sized, thin-walled cysts are observed (arrow), and the diagnosis is confirmed by excisional biopsy. pSS, primary Sjögren’s syndrome; ILD, interstitial lung disease; GGO, ground glass opacity.
NSIP was the most prevalent chest CT pattern in patients with pSS-ILD (51%), followed by UIP (22%) and OP (13%) (Fig. 2B). Histopathology was available for 24 patients with pSS. Biopsy findings were largely consistent with chest CT findings, with some differences: a case of a lung mass on chest CT was fibrotic interstitial pneumonitis in the biopsy, a case of OP on chest CT was UIP on biopsy, and two cases of NSIP on chest CT was OP on biopsy.
PFT results at the diagnosis of ILD were available for 76 patients (96%); 54% of patients with pSS-ILD showed a restrictive functional pattern (FVC < 80%) and 67% of patients showed reduced diffusion capacity (DLCO < 70% of predicted).
During the follow-up period, 14 patients (1.1%) died; including 7/79 with ILD and 7/1,227 without ILD (p < 0.001). The 5-year survival rates were 91.8% and 99.7% for SS patients with and without ILD, respectively. The mortality rate was higher in patients with ILD (log-rank p < 0.001, Fig. 2C).
Treatment of pSS-ILD at diagnosis
Upon diagnosis of ILD, 32 patients (40.5%) initiated treatment for ILD; 41% of them were treated with high-dose glucocorticoids (at a prednisolone equivalent ≥ 30 mg/day) and an immunosuppressant combination. Azathioprine was the most frequently prescribed immunosuppressant (9 patients, 28%), followed by monthly intravenous cyclophosphamide (5 patients, 16%) and MMF (1 patient, 3.1%). The other 59% were treated with glucocorticoids alone. One patient received pirfenidone in combination with high-dose glucocorticoid and MMF.
More patients with OP on baseline chest CT initiated treatment for ILD. Other chest CT patterns and findings did not differ between treatment initiators and non-initiators. However, the baseline lung function was significantly reduced in treatment initiators (Table 2). Additionally, a higher percentage of treatment initiators reported dyspnea (62.5% of initiators versus 38.3% of non-initiators, p = 0.06). Patients receiving treatment showed higher white blood cell counts and NLRs than those without treatment at diagnosis of pSS-ILD. However, levels of C-reactive protein (CRP) and immunoglobulin G did not differ between the two groups.
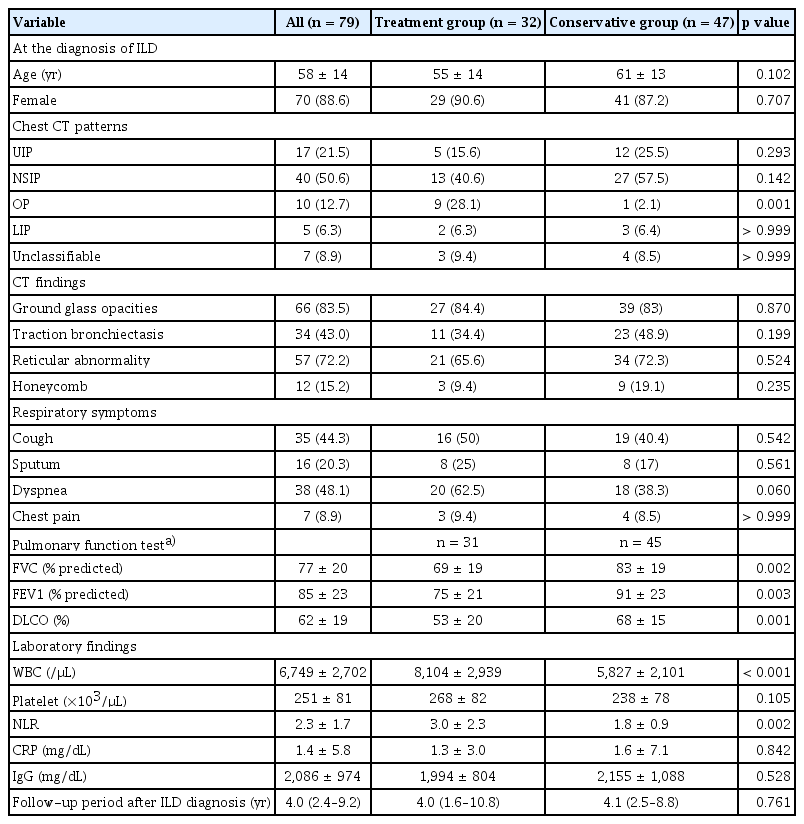
Comparison of baseline clinical characteristics of ILD patients
Long-term clinical course of pSS-ILD
The median follow-up period after ILD diagnosis was 4 years (interquartile range [IQR], 2–9). AEs occurred in 15 patients (19%) after a median of 34 months (IQR, 16–66). Six were reported in patients who did not initiate treatment after diagnosis of ILD. Patients with AE were treated with high-dose glucocorticoids (10 patients, 67%), azathioprine (4 patients, 27%), cyclophosphamide (3 patients, 20%), and MMF (2 patients, 13%). Pirfenidone was prescribed to 2 patients and nintedanib was to a patient.
During the observation period, 20 patients were hospitalized due to respiratory symptoms 56 times. Of these, 17 hospitalizations in 11 patients were due to respiratory tract infection, and 27 hospitalizations in 15 patients were due to AE. The other hospitalizations were associated with pulmonary hypertension.
Patients who experienced an AE had higher NLR compared with those without AE (Fig. 3A). The NLR gradually increased from 6 months before AE. In patients who demonstrated improvement after initial treatment and did not experience an AE, the NLR decreased and was maintained in follow-up tests. NLR did not change significantly in stable patients (Fig. 3A). The AUC value for NLR measured 3 months prior to the occurrence of AE exhibited a modest predictive capacity in distinguishing between patients who would experience AE and those who would not (AUC = 0.618, p = 0.029). An optimal discriminatory threshold for NLR was identified at 1.8, yielding a sensitivity of 80%, and a specificity 59%.
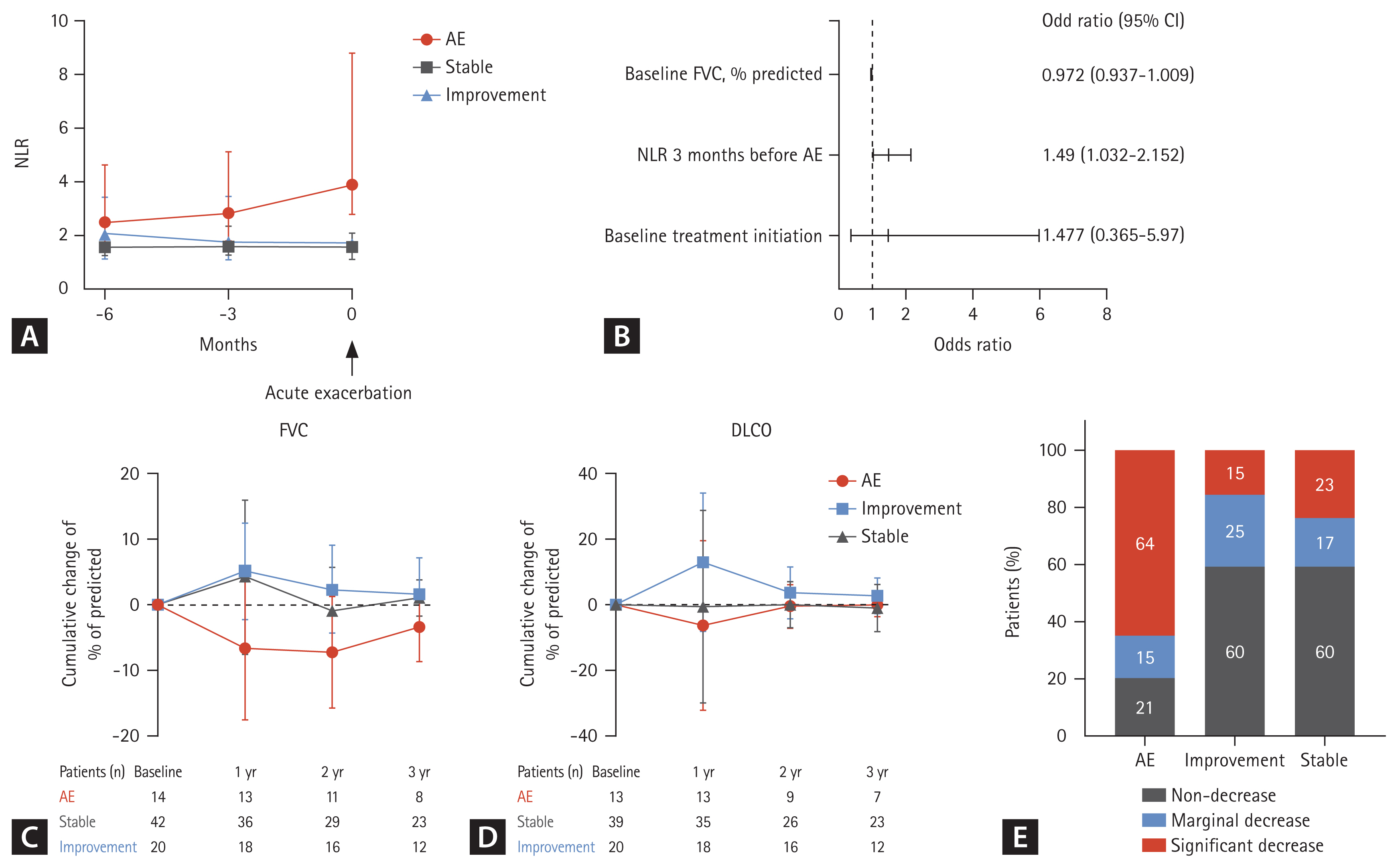
The long-term clinical outcomes of pSS-ILD were grouped into three categories: patients who experienced an AE during follow-up (AE), those who maintained a stable condition without treatment (stable), and those who received treatment from the onset of the disease and did not experience an AE (improvement). (A) Changes in a NLR at 3 months and 6 months before AE. The NLR of patients with AE was compared with those at baseline, 3 months, and 6 months of patients with stable disease and improvement. (B) In the multivariate analysis, adjusted for baseline FVC and the initiation of treatment, the NLR measured three months prior to the occurrence of AE was identified as an independent predictor for AE. (C) Annual cumulative changes in FVC and (D) DLCO, relative to the baseline in pSS-ILD according to clinical outcomes. (E) Proportions of patients with a significant lung function decline according to the clinical course during the observation period. AE, acute exacerbation; NLR, neutrophil-lymphocyte ratio; FVC, forced vital capacity; DLCO, diffusing capacity of the lungs for carbon monoxide; pSS, primary Sjögren’s syndrome; ILD, interstitial lung disease.
To estimate predisposing factors of AE, univariate analyses were performed. Baseline chest CT patterns and treatment for ILD at baseline did not demonstrate any significant associations with AE. In the multivariate analysis adjusted by baseline FVC and and the initiation of treatment, the NLR measured 3 months prior to the occurrence of AE was an independent predictor for AE (OR, 1.49; 95% CI, 1.032–2.152) (Fig. 3B).
In total, 67 patients (85%) had accessible follow-up PFT data. In patients who maintained a stable condition of ILD without treatment (stable), FVC and DLCO did not changed significantly over time. However, in patients who received treatment from the onset of the disease and did not experience any AE (improvement), FVC was increased at 12 months compared to the baseline measurement (mean increase of 5.1% predicted, p = 0.034). In patients who experienced AE, FVC was decreased at 12 months compared to the baseline measurement (mean decrease of 6.2% predicted, p = 0.068) (Fig. 3C). Furthermore, DLCO was increased at 1-year follow-up (mean increase of 13% predicted, p = 0.021) for patients with improvement (Fig. 3D).
Over median 4 years, 20 patients (29%) showed a significant decline in pulmonary function. In patients who experienced AE, 64% showed a significant decline in pulmonary function, while patients with stable disease and those who improved typically demonstrated preserved pulmonary function (Fig. 3E).
In unadjusted analyses, older age at the incidence of pSS-ILD and lower FVC at baseline were parameters for a significant decline in lung function. In multivariate analysis adjusted by lower FVC at baseline, older age at onset of pSS-ILD was an independent risk factor for a decline in lung function (Table 3).
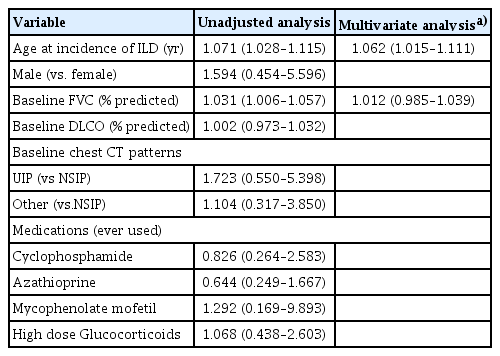
Prognostic factors for a decline in a significant pulmonary function in patients with pSS-ILD
Of 79 patients, 32 (40.5%) underwent echocardiography for screening pulmonary hypertension, and 7 were suspected of having pulmonary hypertension. Two patients received endothelin receptor antagonists after catheterization of the right heart.
In total, 7 patients with pSS-ILD (8.9%) died. The death occurred at a mean of 6.2 ± 5.4 years after the diagnosis of ILD. The major causes of death in patients with pSS-ILD were AE (n = 4), followed by bacterial pneumonia (n = 3). Notably, four deceased patients experienced recurrent episodes, leading to 2–3 readmissions during the follow-up periods, primarily due to AE and/or respiratory tract infections.
DISCUSSION
ILD is one of the most intriguing manifestations of pSS. In this study, 6% of patients with pSS also had ILD, which is the most common extraglandular manifestation associated with mortality in patients with pSS. Over a median observation period of 4 years, AE, a significant decline in pulmonary function, and death occurred in 19%, 29%, and 9% of patients with pSS-ILD, respectively.
ILD was more frequently associated with male pSS patients. This result was consistent with that of a previous study [5]. In contrast, pSS occurs more frequently in females, with a ratio of 1:10 in comparison with males [2]. Male patients with pSS often present with severe manifestations and poor prognosis compared with female patients [17]. Furthermore, patients who developed ILD had a higher age at diagnosis of pSS and were more frequently positive for the anti-Ro antibody. The presence of anti-Ro antibodies is linked to a more active disease in pSS, including ILD [18]. Ro52 (52-kDa protein) is a secondary antigenic target of anti-Ro antibodies in sera of patients with pSS [19]. A Chinese cohort study revealed an association between anti-Ro52 antibodies and the presence of ILD [20]. However, in a more recent study conducted in South Korea, anti-Ro52 antibodies were not associated with the presence or progression of ILD [21].
NSIP is the most common type of pSS-ILD, a finding that is consistent with other studies [4,22,23]. The clinical course of patients with NSIP varies from reversible or self-limited forms to progressive irreversible fibrosis [13]. Compared with a previous study that reported a significant decline in lung function in 50% of patients with pSS-ILD [24], this study observed a decrease in lung function in 29% of patients. While previous studies evaluated patients seen in the pulmonary departments for more advanced disease [8,24,25], pSS-ILD patients in this study were identified in the rheumatology department. These patients have a milder form of ILD and show a higher prevalence of anti-Ro antibodies, which could potentially be linked to a selection bias. Some pSS-ILD patients are diagnosed with ILD before being diagnosed with pSS and often present as seronegative [26]. This scenario can pose challenges in distinguishing pSS-ILD from an unclassifiable ILD or IPF. None of the chest CT patterns in this cohort were related to patient outcomes in pSS-ILD, which is consistent with the findings of a previous study [8]. However, other studies have shown that patients with UIP pattern on chest CT had a poorer clinical course and survival [10,24].
The 5-year mortality rate of pSS-ILD has been reported as 10–20% [8,24,27], and was 9% in the present cohort. Compared with the reported 5-year mortality rates of ILD in connective tissue disease (CTD) ranging from 40–60% [18,28,29], the prognosis of ILD in pSS is favorable among CTD-ILD.
AE was a significant cause of mortality and decline in pulmonary function, suggesting the importance of predictive factors of AE in ILD. Recent studies identified Krebs von den Lungen-6 (KL-6) as an important biomarker that is directly associated with the pathogenic process of ILD [8,30]. KL-6 is secreted by type II alveolar pneumocytes and bronchial epithelial cells in response to cellular damage [31,32]. A higher KL-6 level is also a significant predictor of mortality in patients with pSS-ILD [30]. However, serum KL-6 measurements are often unavailable for routine clinical use. In this study, KL-6 was measured at diagnosis in only 5 patients with SS-ILD.
The NLR is easily and inexpensively measured from a CBC. The NLR is associated with the development of ILD and DLCO in systemic sclerosis patients [33]; mortality rate in patients with dermatomyositis/polymyositis-ILD [34]; and lung function, progression, and mortality of IPF [35]. Furthermore, the NLR demonstrated a pattern of gradual increase several months before the onset of AE in patients with SS-ILD. The reason for the increase in NLR before the onset of AE of ILD is uncertain. The increased percentage of neutrophils in the BAL of patients with IPF has been reported to be associated with a poor outcome [36]. We hypothesized that the elevation in NLR is associated with neutrophilic inflammation in the pathogenesis of ILD. Moreover, we assert that the increase in NLR 3 months prior to the occurrence of AE is independent of overt infection. To assess predictor for AE, we excluded patients clinically diagnosed with infection and those who initiated antibiotics within 6 months before AE, as well as those with a CRP level greater than 10 mg/L. This finding suggests that the NLR may serve as a potential predictor of AE of ILD, although further validation is warranted. The NLR can increase due to factors other than the AE of ILD [37]. If the NLR rises during follow-up, it is necessary to exclude infection such as pneumonia, and to conduct short-term follow-up.
This study has several limitations. First, therapeutic regimens depended on the discretion of the attending physicians and were not consistent across the patients. This variability may have affected clinical course and outcome. Second, some key data, including PFT and chest CT findings, were missing. Some tests were not performed due to patient death or respiratory distress. Thus, the decline in pulmonary function may be underestimated in this study. Third, the extent of ILD on chest CT was not measured, although it can be an important prognostic factor of SS-ILD. Moreover, while HRCT is the optimal imaging modality for ILD detection, a subset of patients in this study did not undergo HRCT at diagnosis. As a real-world observational study, not all patients with pSS underwent HRCT for screening ILD. This variability in imaging modalities may affect the estimated prevalence of ILD in pSS. Fourth, due to the retrospective design of the study, demographic information, including smoking, alcohol consumption, and occupational environment, all which may be associated with lung function decline, were not evaluated. Lastly, the pSS population might be underrepresented in the patient selection algorithm based on ICD codes and laboratory test results. Notably, anti-centromere positive patients were excluded from this study, however, they did not exhibit features of systemic sclerosis [38,39]. Furthermore, we identified patients with ILD among the pSS patients who underwent chest CT. Consequently, selection bias related to the indication for chest CT may be present. Despite these limitations, this study included all pSS patients treated in three university-affiliated hospitals who were diagnosed with pSS by rheumatologists and showed the long-term clinical course and outcomes in a large number of patients with pSS-ILD. To date, this study is the first to show that elevation of the NLR several months before an AE could be a predictive factor of AE development.
In conclusion, ILD is not uncommon (6%) in patients with pSS. Male and elderly patients with pSS were associated with ILD. pSS-ILD has a relatively good prognosis. However, 29% of patients experienced a significant decline in pulmonary function, and 9% of patients died, which was mostly associated with AE. This study suggests a high NLR is a predictor of AE in patients with pSS-ILD.
KEY MESSAGE
1. ILD is found in 6% of in patients with pSS.
2. Older age at diagnosis of pSS-ILD is associated with a significant decline in pulmonary function.
3. The NLR may serve as a potential predictive factor for AEs in pSS-ILD patients.
Acknowledgments
We would like to express sincere gratitude to Bo Mi Gil, the thoracic radiologist at Bucheon St. Mary’s Hospital, for invaluable assistance for this study.
Notes
CRedit authorship contributions
Jung Hee Koh: conceptualization, methodology, resources, investigation, data curation, formal analysis, writing - original draft, writing - review & editing, project administration; Youngjae Park: resources, data curation, formal analysis, writing - review & editing; Jennifer Lee: conceptualization, resources, investigation, data curation, writing - review & editing, supervision; Howook Jeon: conceptualization, investigation, data curation, writing - review & editing; Su-Jin Moon: conceptualization, resources, investigation, data curation, validation, writing - review & editing; Yong Hyun Kim: conceptualization, resources, investigation, writing - review & editing; Jun-Ki Min: conceptualization, resources, writing - review & editing, supervision; Sung-Hwan Park: conceptualization, resources, writing - review & editing, supervision; Seung-Ki Kwok: conceptualization, investigation, data curation, formal analysis, writing - original draft, writing - review & editing, visualization
Conflicts of interest
The authors disclose no conflicts.
Funding
None