Glial cell line-derived neurotrophic factor and its role in attenuating renal fibrosis: a review
Article information

Abstract
Chronic kidney disease is estimated to affect approximately 10 to 15% of the Chinese population. Renal fibrosis is characterized by progressive extracellular matrix deposition in the kidney parenchyma with eventual tissue scarring and inevitable deterioration of renal function. Vascular rarefaction, glomerulosclerosis, interstitial inflammation, and fibrogenesis are associated with or contribute to renal fibrosis. Recent studies have revealed that glial cell-derived neurotrophic factor (GDNF) is involved in kidney morphogenesis and amelioration of renal injury. Ideal therapies targeting the pathogenesis of renal fibrosis should have the potential to inhibit glomerular and tubulointerstitial fibrosis by targeting multiple pathological events. GDNF plays a unique role in both renal development and improvement of renal fibrosis, and GDNF kidney receptors and signaling pathways can ameliorate renal apoptosis and inflammation. Our work contributes to the establishment of GDNF as an emerging therapy that can increase the effectiveness of currently used interventions to improve renal fibrosis. This literature review focuses on the important role of GDNF in renal development and its relationship with renal fibrosis.
INTRODUCTION
Kidney diseases cause serious health problems worldwide, progressing from the early stages of chronic kidney disease (CKD) to end-stage renal disease [1]. Morbidity and mortality rates are very high, and many patients require dialysis or kidney transplantation [2–4]. Renal fibrosis is one of the most common causes of CKD [5]. The concept of CKD is constantly being refined in response to new research findings. CKD is currently defined as a 3-month period of gradual loss of normal kidney function or kidney damage involving structural or functional renal abnormalities with or without a low estimated glomerular filtration rate [5,6]. In 2017, approximately 1.2 million people worldwide were projected to die of CKD, positioning CKD as the 12th most prevalent cause of death globally. Moreover, 35.8 million people were estimated to experience disability-adjusted life years [7,8]. Various abnormalities such as tubulointerstitial fibrosis, loss of renal parenchyma, glomerulosclerosis, and inflammatory cell infiltration are consequences of renal fibrosis [9]. These pathologic outcomes are frequently the result of intricate underlying biological processes such as epithelial–mesenchymal transition (EMT) activation, fibroblast activation, monocyte/macrophage infiltration, and cellular death [9,10]. Following the first insult, resident kidney cells become activated, resulting in the synthesis and secretion of proinflammatory chemotactic cytokines such as transforming growth factor-1 (TGF-1) and tumor necrosis factor [11]. These cytokines cause inflammatory monocytes/macrophages and T cells to infiltrate the damaged areas, and then these cells in the glomerulus or interstitial space become activated and create reactive oxygen species as well as inflammatory and fibrogenic cytokines. Myofibroblasts are generated by the EMT, which stimulates mesangial cells, fibroblasts, and tubular epithelial cells [12,13]. As a result, a large number of extracellular matrix (ECM) components are produced, which are deposited in extracellular compartments and are resistant to destruction. Progressive deposition alters the kidney architecture, leading to collapse of the renal parenchyma and loss of kidney function [14,15]. TGF-1 is a member of the TGF-β superfamily, which includes glial cell-derived neurotrophic factor (GDNF), a strong neurotrophic survival factor [16]. GDNF was first discovered to be a survival factor for noradrenergic, dopaminergic, and spinal motor neurons. Studies have demonstrated its usefulness for treating Parkinson’s disease and other neurological diseases [17–19]. The ligands of this family tend to bind to TGF-β receptors and cause the recruitment and activation of Smad family transcription factors, which in turn influence gene expression. The Smad protein binds to the rearranged during transfection (RET) proto-product, an oncogene that belongs to the receptor tyrosine kinase (RTK) subfamily and is required for the development of the enteric nervous system and kidney [20]. After GDNF knockout in one study, mice demonstrated impairment in both ureteric bud (UB) formation and branching during metanephros development; this was the discovery of non-neuronal GDNF functions [21]. The mice did not survive long after birth because of renal agenesis. Thus, the involvement of GDNF in renal development and illness was revealed.
GDNF was first purified from B49 glial cells. It is an effective neurotrophic factor that can protect nigral dopaminergic neurons [22]. A recent study showed that human amniotic fluid-derived stem cells (AFSCs) have enhanced regeneration ability. In addition, these cells exhibit improved survival, differentiation, and secretion of regeneration factors, particularly in the presence of GDNF [23]. GDNF-modified adipose-derived mesenchymal stem cells (GDNF-AMSCs) inhibit renal inflammation and oxidative stress in mice with unilateral ureteral obstruction (UUO). They also improve peritubular capillary (PTC) injury, promote angiogenesis, and delay the progression of interstitial fibrosis to some extent. Moreover, GDNF-engineered human AFSCs have been shown to play a protective role in kidney injury [24]. GDNF has both dopaminergic and nephrogenic renal morphogenetic actions [25]. However, little is known about the detailed mechanisms underlying these processes.
In this review, we objectively evaluate recent studies on the role of GDNF in renal function. We also extrapolate the mechanism by which GDNF protects the damaged kidney.
ROLE OF GDNF IN KIDNEY DEVELOPMENT
Mammalian embryos contain a pair of mesonephric organs called the Wolffian ducts (WDs), which are induced by signals from metanephric mesenchyme (MM) cells to develop into an epithelial growth called the UB [26,27]. This is the first step in the development of the mammalian (metanephric) kidney. The UB grows and branches from the connecting segment to its entrance into the bladder, eventually giving rise to the tree-like collecting system of the kidney [28]. The induction and proper growth of the UB are critical for normal formation of the kidneys because the subsequent elongation and branching of the UB dictate the renal architecture, which is a fundamental determinant of kidney function. This occurs through the induction of the mesenchymal-to-epithelial transition of the MM, which governs the spatial arrangement of nephrons [27,28]. According to Song et al. [29], GDNF plays a significant role in renal morphogenesis. RTK-activated signaling is a critical regulator of kidney development. The two primary inducers of RTK in the developing kidney are RET (induced by GDNF) and fibroblast growth factor (FGF). RET and GDNF family receptor alpha-1 (GFRA1) receptors and coreceptors are responsible for critical tasks [30,31]. GDNF plays several roles during kidney embryogenesis; it is involved in budding of the ureteric duct from the WD, branching of the ureteric epithelium inside the MM, and in the production of new nephrons [32]. Exogenous GDNF increases the number of ureteric branches and developing nephrons in metanephric culture [32,33]. The number of nephrons that develop during embryogenesis is likewise controlled by GDNF. Deletion of one GDNF allele reduces the number of nephrons in adult kidneys, possibly due to impaired morphogenesis of UB branching [33]. Towers et al. [34] discovered that GDNF promotes UB outgrowth and improves UB cell survival in vitro. Similarly, Pepicile et al. [35] found that GDNF promotes ureter tip proliferation in mice. This results in an increase in the number of ureter tips as well as growth and fusion of neighboring tips, with some tips growing toward the GDNF source. In the late 1990s, knockout investigations established that homozygous null mutants for GDNF, RET, and GFRA1 showed bilateral renal agenesis and died shortly after delivery [36–38].
GDNF RECEPTORS IN THE KIDNEY
RET
As noted above, GDNF signaling through its RET receptors (GDNF/RET signaling pathway) is essential for UB growth in the developing kidney. Although the exact process is unknown, recent research suggests that a combination of localized GDNF expression via the MM and many types of negative regulation result in the correct location of the initial budding event from the WD. GDNF continues to promote the branching process of the UB [39]. However, it does not offer the positional information needed to establish the pattern of UB growth and branching because the synthesis position can be significantly modified with no influence on kidney development [40]. Although GDNF-driven proliferation is necessary for the creation and expansion of the UB tip, cells lacking RET cannot contribute to this specialized epithelial region [41]. The early expansion of the UB is a crucial phase in the development of the urogenital system. Neither the ureter nor the kidney develops when this stage does not occur (resulting in renal agenesis) [42]. An ectopic ureter fails to effectively connect to the bladder if it is positioned incorrectly. As a result, urinary system anomalies such as megaureter, vesicoureteral reflux, and ureteroceles are common birth malformations. RET mutations are reportedly responsible for 40% of cases of renal agenesis in humans, whereas GDNF mutations are responsible for 5 to 10% [43].
GFRA1
GFRA1 is also known as RET ligand 1 or TGF-β-related neurotrophic factor receptor 1. It belongs to the GDNF receptor subfamily and is a newly discovered glycosylphosphatidylinositol-linked cell surface receptor [44]. The RET signaling pathway is initiated when GFRA1 facilitates attachment and stimulation of the RET RTK. The survival, differentiation, and migration of peripheral and central neurons depend on GFR-mediated signaling. GFRA1 also plays a role in the inflammatory response in some cancers and in the development of the enteric nervous system and kidneys [45–47].
RENOPROTECTIVE EFFECT OF GDNF AGAINST RENAL FIBROSIS
Renal fibrosis is common in patients with advanced CKD. The most common pathogenic characteristics of renal fibrosis are hypoxia, oxidative stress, inflammatory cell infiltration, fibroblast activation, and ECM accumulation. This causes the degradation of kidney parenchyma, which manifests as tubular atrophy, capillary loss, podocyte depletion, and end-stage renal disease [48]. GDNF was originally reported to be a potential therapeutic agent for the treatment of neurodegenerative diseases before many studies revealed its further roles in treating renal fibrosis and other anomalies [49,50]. One study demonstrated that kidney agenesis or dysgenesis in GDNF−/− mutant mice may result from the promotion of GDNF in ureteric branching, suggesting that GDNF may acts as a morphogen in kidney development [36]. GDNF expression is associated with epithelial differentiation of the nephrogenic mesenchyme in the kidney, and the absence of this gene results in renal aplasia or severe hypodysplasia [21,51]. Therefore, GDNF as a trophic factor has great promise for its use in organogenesis. GDNF is a tissue morphogen that enhances the migration and differentiation of stem cells and reverses kidney injury [52]. Some authors have speculated that pretreatment of AMSCs with GDNF may better alleviate renal fibrosis by strengthening the ability of AMSCs to differentiate into endothelial cells [23,53]. GDNF has therapeutic effects on mesenchymal stem cells, which have emerged as ideal cell candidates for functional and structural restoration of injured kidneys. It can help ameliorate kidney disease, and the hypothesized mechanism involves the transfer of endogenous molecules through paracrine factors to apprehend injured cells. However, the precise mechanism remains unclear. GDNF may activate an angiogenesis pathway in surviving PTCs after injury has occurred, leading to activation of the sirtuin-1 (SIRT1)/endothelial nitric oxide synthase signaling pathway. By inhibiting cell apoptosis and fibrosis, SIRT1 shows cytoprotective effects and inhibits inflammation in the kidney, thus contributing to the maintenance of renal homeostasis. Its downregulation leads to acute and CKDs. Angiogenesis helps regulate the interaction between SIRT1 and endothelial nitric oxide synthase. However, whether angiogenesis and its regulation of SIRT1 are involved in the development of renal fibrosis remains unclear, and the role of GDNF in attenuating this process remains to be defined (Fig. 1).
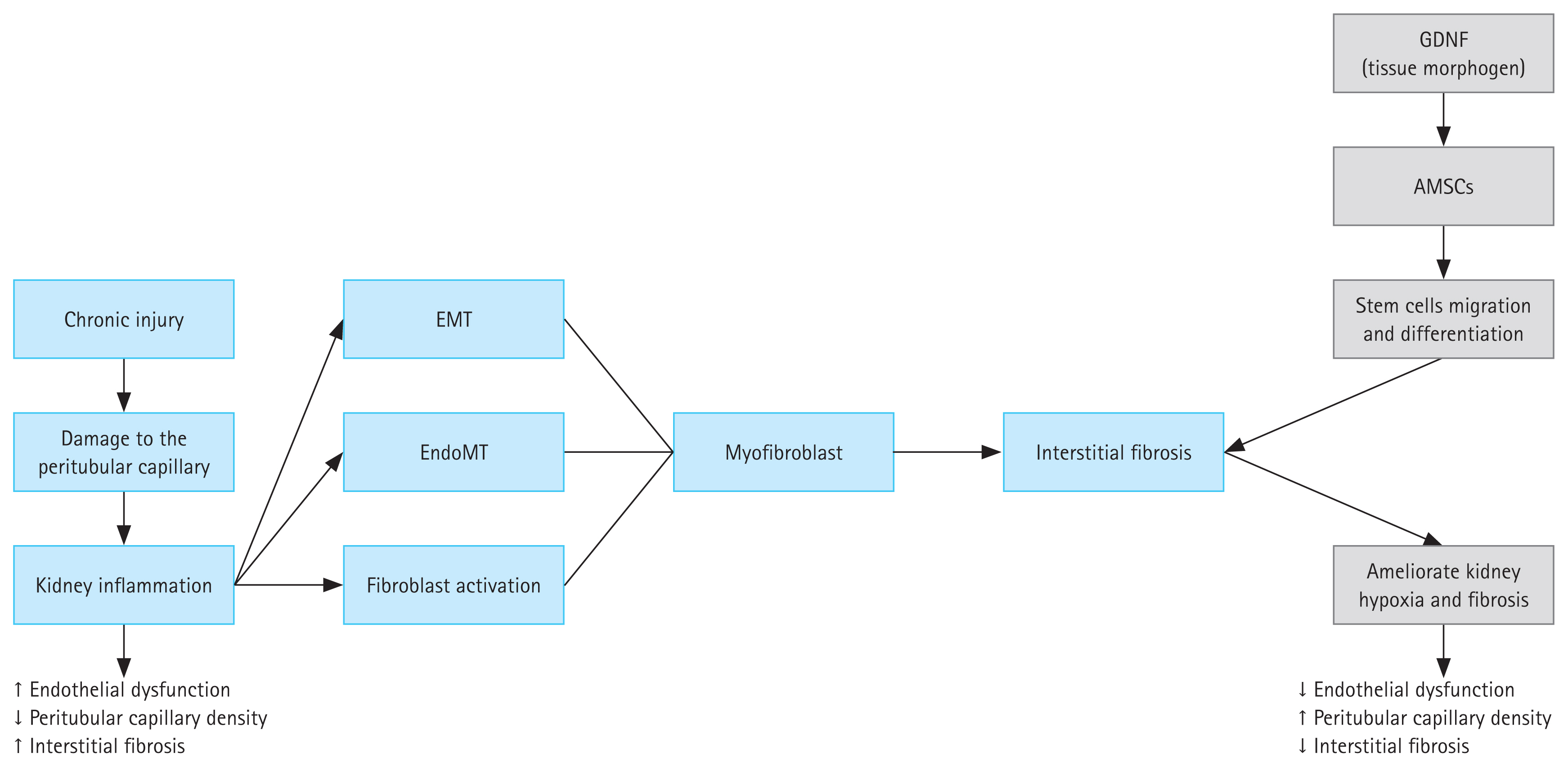
Process of kidney fibrosis caused by chronic injury and the mechanism of action of exosomes derived from GDNF stem cells in the amelioration of kidney fibrosis. Damage to the glomerulus can be caused by many different chronic injuries, leading to endothelial dysfunction. As a response to this inflammatory process, renal tubular cells undergo EMT. During this transformation, these cells exhibit fibroblast-like behaviors, including the generation of collagen. The fibroblasts accumulate in the ECM and damage the kidney tubules and blood vessels, leading to renal interstitial fibrosis. GDNF is a tissue morphogen that can enhance the migration and differentiation of stem cells and reverse kidney injury. Thus, GDNF maintains renal blood flow and effectively suppresses the induction of myofibroblast progression. Transplantation of GDNF can help ameliorate kidney hypoxia and inhibit the progression of renal fibrosis. AMSC, adipose-derived mesenchymal stem cell; ECM, extracellular matrix; EMT, epithelial–mesenchymal transition; EndoMT, endothelial-to-mesenchymal transition; GDNF, glial cell-derived neurotrophic factor.
Cellular events in renal fibrogenesis
Several biological processes, such as EMT, monocyte/macrophage infiltration, T-cell infiltration, mesangial and fibroblast activation, and cell death, are important events that lead to histological manifestations of renal fibrogenesis [54]. Renal fibrogenesis involves a variety of cell types that work together to cause and maintain renal fibrosis on a large scale. Almost all kidney cells play a role in the pathogenesis of renal interstitial fibrosis, including fibroblasts, tubular epithelial cells, podocytes, vascular smooth muscle cells, pericytes, macrophages, and lymphocytes [55]. Following the first insult, injured resident kidney cells become activated, resulting in the synthesis and secretion of proinflammatory chemotactic cytokines. These cytokines act as directional signals, resulting in the movement of inflammatory monocytes/macrophages and T lymphocytes to the wounded tissue [56]. Those that infiltrate the glomerulus or interstitial space (depending on the site of injury) therefore become activated and produce reactive oxygen species as well as inflammatory and fibrogenic cytokines, which stimulate mesangial cells, fibroblasts, and tubular epithelial cells [57]. Then they undergo phenotypic transition and activation, creating a considerable amount of ECM components [58]. ECM proteins are resistant to degradation and are deposited in extracellular compartments. Progressive deposition alters the kidney architecture, leading to collapse of the renal parenchyma and loss of kidney function.
Molecular events in renal fibrogenesis
The TGF/Smad signaling system and its downstream Smad signaling pathway play important roles in renal fibrogenesis [59]. Several studies have shown that TGF-β is the critical mediator in CKD associated with increasing renal fibrosis. TGF-β appears to be involved in numerous biological functions, including cell proliferation, differentiation, apoptosis, autophagy, and ECM synthesis [60]. Its upregulation has been found in nearly every type of CKD, both in animal models and in humans. It may induce myofibroblastic activation or transition in mesangial cells, interstitial fibroblasts, and tubular epithelial cells in vitro, resulting in matrix-producing fibrogenic cells [61]. Renal fibrosis is caused by the expression of exogenous TGF-β, either in vivo or in transgenic mice. Multiple techniques that inhibit TGF-β decrease renal fibrotic lesions and prevent the gradual loss of kidney function [62]. Smad3 is strongly triggered during fibrogenesis, and this is followed by loss of the inhibitory Smad7 via a ubiquitin E3/ligase-dependent degradation pathway [63]. The symmetrical shift between Smad3 and Smad7 causes myofibroblasts to congregate and activate, overproduce ECM, and reduce ECM breakdown in the damaged kidney [64]. Nonetheless, there are other TGF-β/Smad signaling pathway downstream effectors. TGF-β is utilized by Smad2, which activates its renal protective effect by inhibiting Smad3 [65]. In this way, TGF-β/Smad signaling plays a critical role in reducing renal fibrosis.
GDNF AND ITS SIGNALING PATHWAYS
GDNF requires numerous mediators in distinct signaling pathways to carry out its functions in vivo and in vitro. In this review, we examine the phosphoinositide 3-kinase (PI3K)/Akt and mitogen-activated protein kinase (MAPK)/extracellular signal-regulated kinase (ERK) pathways and how they relate to GDNF-mediated kidney protection.
PI3K/AKT signaling
PI3K and Akt or protein kinase B form the P13K/Akt signaling pathway. The Akt/protein kinase B family of protein kinases are serine/threonine protein kinases that regulate growth, metabolism, proliferation, survival, transcription, protein synthesis, and other cellular functions [66]. They function as primary signal transducers after PI3K activation. Akt has three isoforms (Akt1, Akt2, and Akt3) with diverse functions in different organs. Growth factors such as insulin-like growth factor-1 receptor and the epidermal growth factor family of receptors, as well as TGF-β, activate serine/threonine kinases [67].
Effect on renal development
The role of GDNF and the PI3K/Akt signaling pathway in renal morphogenesis has been extensively described. As previously mentioned, GDNF, a member of the TGF-β superfamily of growth factors, causes the UB to develop from the WD via the RET RTK and its coreceptor GFRA1. RTKs, such as RET and FGF receptors, can activate a variety of intracellular signaling pathways (including PI3K/Akt) upon binding of their ligands. Among these signaling pathways, PI3K/Akt signaling appears to be critical for GDNF-dependent expansion of the UB [68].
Effect on ameliorating renal injury
Once activated, the PI3K/Akt signaling pathway effectively prevents kidney damage. Fibroblast stimulation causes cell proliferation and an increase in ECM deposition, resulting in the development of interstitial fibrosis. Different cytokines and growth factors involved in proliferation, such as TGF-β, epidermal growth factor, and platelet-derived growth factor, trigger intracellular signaling pathways culminate on Akt proteins [69]. Inhibition of PI3K/Akt suppresses fibronectin and collagen type 1 production [70]. Thus, Akt activation might be linked to increases in ECM synthesis following renal damage. TGF-β, a profibrotic cytokine, is involved in the initiation and maintenance of fibrosis and has been shown to stimulate fibroblast proliferation in response to Akt activation [71,72]. It also increases cellular hypertrophy, particularly of mesangial cells, by regulating PI3K/Akt. TGF-β-induced hypertrophy and expression of plasminogen activator inhibitors are thought to be controlled by PI3K-activated Akt, which contributes to the abundance of many matrix proteins [73,74]. The EMT is characterized by the loss of epithelial cell properties and the development of ECM-producing myofibroblasts. Multiple stimuli, such as the expression of TGF-1, advanced glycation end products, and angiotensin II [75], can cause tubular EMT. Activation of the PI3K/Akt pathway has been linked to TGF-1-induced renal EMT in vitro and in vivo, resulting in renal fibrosis [76]. PI3K/Akt signaling can effectively control cell survival (inhibit apoptosis) by directly inhibiting pro-apoptotic proteins including BAD and FOXO via RTK activation by GDNF [77,78].
MAPK/ERK signaling
The MAPK/ERK pathway, also known as the Ras/Raf/MEK/ERK pathway, is a protein chain within the cell that transmits a signal from the cell’s surface receptor to the nucleus DNA. Many proteins are involved in the pathway, including MAPK (formerly known as ERK), and these proteins communicate with one another by adding phosphate groups to an adjacent protein [79]. MAPKs are a family of serine/threonine protein kinases that are involved in many essential cellular functions, including proliferation, differentiation, motility, the stress response, apoptosis, and survival [80]. Three families have been identified: p38 MAPK, ERK, and Jun kinase (JNK/SAPK). Stimulation of tyrosine kinase receptors causes MAPKs to be activated in a multistep process in the MAPK/ERK signaling pathway. Activated Ras activates Raf kinase, which then phosphorylates and promotes MEK (MEK1 and MEK2). MEK stimulates and phosphorylates ERK [79].
Effect on renal morphogenesis
One of the most critical receptors involved in kidney development is RTK. GDNF-induced RET signaling and FGF signaling are the most important inducers of RTK signaling in the developing kidney. Those growth factors tend to bind to their associated receptors, resulting in receptor dimerization, autophosphorylation, and activation of intracellular signaling pathways such as the MAPK/ERK pathway [81,82]. MAPK signaling is required by various cell types in the developing kidney. MAPK signaling is primarily important in the morphogenesis of UB branching and in determining the numbers of nephrons that develop, as evidenced by a decrease in UB tip cell proliferation and aberrant branch development when MEK proteins are inhibited [83–86].
Effect in ameliorating renal fibrosis
JNK is one of three well-known MAPK pathways that control activities such as cell proliferation, differentiation, migration, and apoptosis [87]. JNK activation is a common response to many stresses in tubular epithelial cells. Ma et al. [50] used UUO to investigate the role of JNK signaling in renal interstitial fibrosis. First, stretch-induced damage to tubular epithelial cells caused rapid development of interstitial fibrosis in the UUO model. Following UUO, JNK signaling was found in both dilated and atrophic tubules as well as in tubules with a normal shape. Treatment with a JNK inhibitor (CC-401) effectively prevented JNK signaling and provided significant protection against interstitial fibrosis in terms of myofibroblast formation and collagen deposition, and it lowered TGF-1 and connective tissue growth factor mRNA levels.
ROLE OF GDNF IN RENAL FIBROSIS
Renal fibrosis is a complex process that includes macrophage/lymphocyte infiltration, podocyte apoptosis, accumulation of activated fibroblasts, EMT, and enhanced ECM deposition, among other processes [88]. Therefore, an ideal therapy should be able to suppress tubular and glomerular interstitial fibrosis by targeting several events along the pathogenic process. Although GDNF plays an important role in kidney development, it has also been shown to help with renal fibrosis [53].
Li et al. [24] examined whether transplanting GDNF-modified AFSCs for the treatment of renal interstitial fibrosis is more effective than transplanting unmodified AFSCs. GDNF-modified AFSCs significantly reduced oxidative stress and inflammation and regulated the protein levels of PTCs, vascular endothelial growth factor (VEGF), hypoxia-inducible factor-1 (HIF-1), and TGF-1. The GDNF gene enhanced the ability of AFSCs to control inflammation and oxidative stress, which helps alleviate renal fibrosis. They also found that transplanting AFSCs into mice with ureteral ligation boosted tubular epithelial cell proliferation and reduced renal interstitial fibrosis. Anti-inflammatory and anti-oxidative pathways in AFSCs were found to help mice recover more rapidly from UUO.
Wang et al. [89] investigated whether GDNF improves the anti-inflammatory effects of human AMSC-based therapy in renal interstitial fibrosis. They focused on how AMSCs work biologically and whether the GDNF gene can help stem cells to become more anti-inflammatory. Interleukin (IL)-10, IL-4, inducible nitric oxide synthase, and tumor necrosis factor mRNA expression levels were measured via quantitative reverse-transcription polymerase chain reaction (qRT-PCR). Two M2 macrophage markers were discovered using flow cytometry (CD163 and CD206). UUO models were established in nude mice, with six mice in each group. IL-6, TGF-1, smooth muscle actin, and cyclooxygenase 2 were subsequently evaluated via Western blotting. In all groups, pathological changes in the kidneys were observed via Masson staining and hematoxylin and eosin staining. All of the preceding observations led to the conclusion that AMSCs can cause M1 macrophages to change into M2 macrophages and that GDNF can enhance this transformation. Furthermore, AMSC therapy improved renal function and reduced interstitial fibrosis in the UUO model.
Wang et al. [90] examined whether GDNF-AFSCs could reduce apoptosis in the mouse renal tubular epithelial cell (mRTECs) and improve renal dysfunction. They used an mRTEC hypoxia/reoxygenation (ischemia/reperfusion) model. In a Transwell system, damaged mRTECs were cocultured with GDNF-AFSCs. The expressions of FGF and hepatocyte growth factor mRNA were measured via qRT-PCR. The levels of superoxide dismutase, malondialdehyde, and reactive oxygen species were also measured. Bax, Bcl-2, Gp91-phlox, and nitrotyrosine expression were examined via Western blotting. The GDNF-AFSC-treated groups showed an increase in growth factor expression and a considerable counteraction of oxidative stress. Hypoxia/reperfusion-induced apoptosis was also reduced in mRTECs.
Some of the factors that contribute to renal disease progression, which can be affected by chronic tissue hypoxia and PTC rarefaction stemming from microvascular injury, can easily accelerate interstitial kidney fibrosis [91,92]. Hypoperfusion of PTCs can induce hypoxia and increase the progression of tubulointerstitial fibrosis [93]. Chen et al. [94] investigated whether GDNF-AMSC-exos could alleviate kidney hypoxia and fibrosis in UUO models. They specifically examined the expression levels of HIF-1α and VEGF. In a group treated with GDNF-AMSC-exos, there was an increase in VEGF expression, while the expression of HIF-1α was significantly reduced. For summary check Table 1 [24,89,90,94–96]. These results suggest that treatment with GDNF-AMSC-exos can significantly improve kidney hypoxia and thus ameliorate kidney fibrosis.

Summary of the protective roles of GDNF against renal fibrosis and kidney injury
CONCLUSIONS
Tubulointerstitial fibrosis, defined as progressive deposition of damaging connective tissue in the kidney parenchyma, appears to be a destructive process that ultimately leads to renal function decline, regardless of the primary renal disease that causes the initial kidney injury. Despite current CKD treatment choices and efforts to eradicate the condition, further research is required. The use of GDNF is a new modality that has the potential to benefit patient outcomes and complement existing therapies. The importance of GDNF in renal development and the repercussions of its absence during embryogenesis were highlighted in this review. Furthermore, GDNF kidney receptors were discussed, and signaling pathways employed to reduce renal apoptosis, inflammation, and fibrosis were examined. Most studies to date have reported positive effects of GDNF although some have suggested that GDNF might increase the growth of cancerous cells; additionally, high levels of GDNF are associated with pancreatic tumors. A limitation of this review is that few studies have focused on the role of GDNF in the kidney and the mechanism underlying its above-described roles, which limits a more comprehensive understanding. Consequently, few clinical trials have been conducted to assess the effects of GDNF on patients; most have been conducted on stem cells and animal models. Therefore, more studies are needed to gain a better understanding of the safety of GDNF for the kidney.
Acknowledgments
We would like to thank our colleagues at the Department of Nephrology for their support and care.
Notes
CRedit authorship contributions
Yousuf Abdulkarim Waheed: methodology, resources, writing - original draft, writing - review & editing; Wokuheleza Buberwa: methodology, writing - review & editing; Dong Sun: conceptualization, supervision, project administration, funding acquisition
Conflicts of interest
The authors disclose no conflicts.
Funding
This study was supported by funding from the National Natural Science Foundation of China (82270731, 82000703), the Jiangsu Provincial Natural Science Foundation (BK20211054), a project of Qing Lan of Jiangsu Province, a project of Jiangsu Provincial Post Graduate Innovation Plan (KYCX21_2701, KYCX22_2903), the Science and Technology Development Fund of the Affiliated Hospital of Xuzhou Medical University (XYFC2020001; XYFY2020038), the Xuzhou Key R & D Program (Social Development) (KC20160), the Xuzhou Medical Leading Talent Training Project (XWRCHT20210038), and the Beanstalk Talent of the Affiliated Hospital of Xuzhou Medical University.